Синдромът възниква поради липсата на част от генетичния материал, разположен върху късото рамо на хромозома 11. Премахването на част от генетичния материал се нарича делеция. Изтриването води до поражението на онези функции, които е трябвало да се изпълняват от изгубените гени.
Всички гени, с изключение на някои, които са разположени на половите хромозоми, са представени в два екземпляра. Всеки човек получава една порция гени от майка си и втора идентична част от баща си. Те от своя страна са получили своите двойки гени от своите родители. Генетичният материал се предава от родителите чрез репродуктивни клетки. Половите клетки (яйцеклетка или сперма) са единствените клетки в тялото, които носят само едно копие на генетичен материал. Преди да влезе генетичният материал полова клетка, между две копия на гени, гените се разбъркват и всеки родител поставя генетичен материал в репродуктивната клетка, която е смес от материала, който тя на свой ред е получила от родителите си. Нов животте също ще бъдат разбъркани, преди да бъдат поставени в репродуктивната клетка, за да се създаде следващото поколение. Този процес се нарича кросингоувър. Това се случва между хомоложни области на хромозоми по време на образуването на зародишни клетки. Чрез процеса на кросингоувър гените могат да създават нови комбинации. Това смесване осигурява разнообразието на новите поколения. За какво е? Това е необходимо, за да се осигури променливост на поколенията, в в противен случайщяхме да предадем на нашите деца точни копия на хромозомите, получени от един от нашите родители, променливостта между поколенията би била изключително ограничена, което би направило биологична еволюцияна Земята би било изключително трудно и следователно би намалило шансовете за оцеляване. В момента, в който протичат такива процеси, парче от хромозомата може да се отлепи и да се получи „изтриване“. Делецията е вид мутация. Ако се появи за първи път, тогава такава мутация се нарича de novo мутация (първата, първоначална). В допълнение към мутациите, които са се появили за първи път в тялото, има мутации, които са наследени. Мутация de novo може да бъде предадена на следващите поколения, в който момент вече няма да се нарича мутация de novo.
При синдрома на WAGR част от генетичния код се премахва и няма достатъчно генетичен материал.
В природата има противоположни условия, когато болестта се проявява поради допълнително копие на генетичен материал.
Проявата на WAGR синдром зависи от това кои гени са изключени в резултат на делецията. Съседните гени винаги отпадат. При WAGR генът PAX6 и генът WT1 винаги се губят, което води до типична проява на заболяването. Точковите мутации в гена PAX6 водят до аниридия, а мутациите в WT1 водят до тумор на Wilms. При WAGR няма мутация на тези гени - самите гени отсъстват.
Хората със синдром на WAGRO (буквата O е добавена - затлъстяване) имат увреждане на BDNF гена. Този ген се експресира в мозъка и е важен за живота на невроните. Протеинът, произведен под въздействието на този ген, най-вероятно участва в регулирането на ситостта, жаждата и телесното тегло. Загубата на BDNF най-вероятно е свързана със затлъстяването, което започва през детствопри деца със синдром на WAGRO. Пациентите с WAGRO имат по-голям риск от неврологични проблеми като намалена интелигентност и аутизъм. Не е напълно проучено дали този риск е свързан конкретно със загубата на BDNF гена
Знаем нещо за гените, които са изключени при WAGR синдрома:
WT1
WT1 е ген (туморен ген на Wilms), който секретира протеин, необходим за нормално развитиебъбреци и полови жлези (яйчници при жените и тестиси при мъжете). В тези тъкани протеинът играе роля в клетъчната диференциация и апоптозата. За да постигне всичко това, WT1 функционира, за да регулира активността на други гени чрез свързване на региони на ДНК.
Генът WT1 е необходим за потискане на тумора на Wilms. Съществува вариант на наименованието на гена Wilm's tumor tumor supressor gene1 (ген, потискащ развитието на тумора на Wilms).Неговата мутация или липса води до повишен риск от развитие на тумора именно поради вероятността от участие на този ген при WAGR синдром, че е необходимо постоянно наблюдение на състоянието на бъбреците.
PAX6
PAX6 принадлежи към семейство гени, които играят критична роля в развитието на органи и тъкани по време на ембрионално развитие. Членовете на семейството PAX са важни за нормалното функциониране на различни клетки в тялото след раждането. Гените от семейството PAX участват в синтеза на протеини, които свързват специфични участъци от ДНК и по този начин контролират активността на други гени. Поради това свойство PAX протеините се наричат транскрипционни фактори.
По време на ембрионалното развитие протеинът PAX 6 активира гени, участващи в развитието на очите, мозъка, гръбначния мозък и панкреаса. PAX 6 участва в разработката нервни клеткиобонятелен тракт, които са отговорни за обонянието. В момента функцията PAX 6 по време на вътрематочно развитиеНай-вероятно не е напълно проучен и с течение на времето получаваме нови факти. Веднъж роден, протеинът PAX6 регулира много гени в окото.
Липсата на функция на гена PAX 6 води до проблеми с очите, възникващи след раждането.
BDNF
Генът BDNF кодира протеин, който се намира в мозъка и гръбначен мозък. Този ген играе роля в растежа и узряването на нервните клетки. BDNF протеинът е активен в синапсите в мозъка. Синапсите могат да се променят и адаптират в отговор на опита. BDNF протеинът помага за регулиране на синаптичната променливост, което е много важно за ученето и паметта.
BDNF протеинът се намира в областите на мозъка, които контролират ситостта, жаждата и телесното тегло. Най-вероятно този протеин допринася за тези процеси.
Експресията на този ген е намалена при болестите на Алцхаймер, Паркинсон и Хънтингтън и този ген може да играе роля в реакциите на стрес и заболяванията, свързани с разстройството на настроението. Генът BDNF привлече вниманието на много изследователи. Има изследвания, които изследват активността на протеина BDNF в мозъка в зависимост от физически упражнения, диети, психически стреси други условия. Дейността на този протеин е свързана с умствената дейност и психически състояния, се правят опити да се повлияе нивото му.
Ще съм благодарен да ми посочите за нова информацияотносно този въпрос. Напишете всичко в коментарите.
Забележка:
Думите протеин и протеин са синоними
Е.В. Тозлиян, детски ендокринолог, генетик, д-р Отделен структурно подразделение"Научно-изследователски клиничен институт по педиатрия" Държавна бюджетна образователна институция за висше професионално образование Руски национален изследователски медицински университет на името на. Н.И. Пирогов, Министерство на здравеопазването на Руската федерация, Москва Ключови думи
: деца, синдром на Нунан, диагноза.
Ключови думи: деца, синдром на Нунан, диагностика.
Статията описва синдрома на Нунан (синдром на Улрих-Нунан, турнероиден синдром с нормален кариотип) - рядък вродена патология, се унаследява по автозомно-доминантен начин, е фамилен, но се срещат и спорадични случаи. Синдромът предполага наличието на фенотип, характерен за синдрома на Шерешевски-Търнър при индивиди от женски и мъжки пол с нормален кариотип. Представен от клинично наблюдение. Трудностите на диференциално-диагностичното търсене и липсата на информираност на клиницистите за този синдроми значението на интердисциплинарния подход.
Исторически факти
Необичайният синдром е споменат за първи път от О. Кобилински през 1883 г. (снимка 1).
Най-старият известен клиничен случайСиндром на Нунан, описан през 1883 г. от О. Кобилински
Заболяването е описано през 1963 г. от американския кардиолог Жаклин Нунан, която съобщава за девет пациенти с клапна стеноза белодробна артерия, нисък ръст, хипертелоризъм, умерен спад в интелекта, птоза, крипторхизъм и скелетни аномалии. Д-р Нунан, който практикува като детски кардиологв университета на Айова, забеляза, че децата с рядък типсърдечно заболяване - стеноза на белодробна клапа - характерна физически аномалиипод формата на нисък ръст, шия с форма на крило, широко разположени очи и ниско поставени уши. Момчетата и момичетата бяха еднакво засегнати. Д-р Джон Опиц, бивш ученикНунан е първият, който изковава термина „синдром на Нунан“, за да опише състоянието на деца, които показват признаци, подобни на тези, описани от Нунан. По-късно Нунан написа статия „Хипертелоризъм с фенотипа на Търнър“ и на симпозиум през 1971 г. сърдечно-съдови заболяванияимето "синдром на Нунан" стана официално признато.
Етиология и патогенеза
Синдромът на Noonan е автозомно доминантно разстройство с променлива експресивност (фиг. 1). Генът на синдрома на Noonan се намира на дълго рамохромозома 12. Не може да се изключи генетична хетерогенност на синдрома. Спорадични и семейни формисиндром с автозомно-доминантна форма на наследяване. В семейните случаи мутантният ген се наследява, като правило, от майката, тъй като поради тежки пороциразвитие пикочно-половата системамъжете с това състояние често са безплодни. Повечето докладвани случаи са спорадични, причинени от de novo мутации.

. Автозомно-доминантен тип наследяване
Описаните комбинации от синдром на Noonan с неврофиброматоза тип I в няколко семейства доведоха до предположението възможна връзкадва независими локуса 17q11.2 на хромозома 17. Някои пациенти имат микроделеции в локуса 22q11 на хромозома 22; в тези случаи клинични проявленияСиндромът на Noonan се комбинира с хипофункция на тимуса и синдром на DiGeorge. Редица автори обсъждат участието на предполагаеми гени за лимфогенеза в патогенезата на синдрома поради наличието на лицеви и соматични аномалии, подобни на синдрома на Търнър и високата честота на патологията лимфна система.
Повечето обща причинаСиндромът на Noonan е мутация на гена PTPN11, която се открива при приблизително 50% от пациентите. Протеинът, кодиран от гена PTPN11, принадлежи към семейство молекули, които регулират отговора еукариотни клеткикъм външни сигнали. Най-голямо числомутациите в синдрома на Noonan са локализирани в екзони 3, 7 и 13 на гена PTPN11, кодиращи протеинови домейни, отговорни за прехода на протеина в активно състояние.
Възможните идеи за патогенезата са представени от следните механизми:
RAS-MAPK-path е много важен пътсигнална трансдукция, чрез която извънклетъчните лиганди - някои растежни фактори, цитокини и хормони - стимулират клетъчна пролиферация, диференциация, оцеляване и метаболизъм (фиг. 2). При свързване на лиганда рецепторите на клетъчната повърхност се фосфорилират на места в тяхната ендоплазмена област. Това свързване включва адапторни протеини (напр. GRB2), които образуват конститутивен комплекс с гуанин нуклеотидни обменни фактори (напр. SOS), които превръщат неактивния GDP-свързан RAS в неговата активна GTP-свързана форма. След това активираните RAS протеини активират каскадата RAF-MEKERK чрез серия от реакции на фосфорилиране. В резултат на това активираният ERK навлиза в ядрото, за да промени транскрипцията на целевите гени и да коригира активността на ендоплазмените мишени, за да предизвика адекватни краткосрочни и дългосрочни клетъчни отговори на стимул. Всички гени, включени в синдрома на Noonan, кодират протеини, неразделна част от този път и мутации причиняващи заболяване, обикновено усилват сигнала, преминаващ през този път.
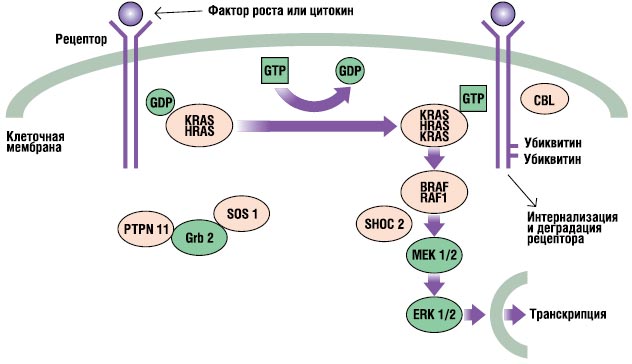
. RAS-MAPK-сигнален път. Сигналите за растеж се предават от рецепторите, активирани от растежен фактор, към ядрото. Мутациите в PTPN11, KRAS, SOS1, NRAS и RAF1 са свързани със синдрома на Noonan, а мутациите в SHOC2 и CBL са свързани с фенотип, подобен на синдрома на Noonan
Клинични характеристики на синдрома на Noonan
Фенотипът на пациентите със синдром на Noonan прилича на синдрома на Turner: къс врат с птеригоидна гънка или нисък растеж на косата, нисък ръст, хипертелоризъм на палпебралните фисури (Фигура 2). Лицевите микроаномалии включват антимонголоидни палпебрални фисури, обърнати надолу външни кантуси, птоза, епикантус, ниско разположени ушни миди, сгъната спирала уши, неправилно захапване, цепнатина на увулата меко небце, готическо небце, микрогнатия и микрогения. Гръдният кош е щитовиден с хипопластични, широко разположени зърна, гръдната кост е изпъкнала в горната част и хлътнала в долната. Около 20% от пациентите имат умерена скелетна патология. Най-често деформация на фунията гръден кош, кифоза, сколиоза; по-рядко - намаляване на броя на шийните прешлени и тяхното сливане, напомнящо за аномалии при синдрома на Klippel-Feil.

. Фенотипове на синдрома на Noonan
Пациентите със синдром на Нунан обикновено имат руса, гъста, къдрава коса с необичаен растеж в темето на главата; тъмни петнавърху кожата, хипертрихоза, дистрофия нокътни плочки, аномалии в никненето и разположението на зъбите, склонност към образуване на келоидни белези, повишена разтегливост на кожата. Една трета от пациентите имат периферен лимфедем; по-често лимфедем на ръцете и краката се среща при деца ранна възраст. Често срещан симптом е патология на зрението (миопия, страбизъм, умерен екзофталм и др.). Забавянето на растежа се среща при приблизително 75% от пациентите, по-изразено е при момчетата и обикновено е незначително. Забавянето на растежа се проявява в първите години от живота, по-рядко се отбелязват леки дефицити на височина и тегло при раждането. От първите месеци на живота се наблюдава намаляване на апетита. Костна възрастобикновено изостава от паспорта.
Характерна особеност на синдрома е едностранен или двустранен крипторхизъм, който се среща при 70-75% от пациентите от мъжки пол, азооспермия, олигоспермия, дегенеративни променитестисите. Независимо от това пубертетът настъпва спонтанно, понякога с известно закъснение. Момичетата често изпитват забавяне на образуването на менструация, а понякога има и нередности менструален цикъл. Фертилитетът може да е нормален при пациенти и от двата пола.
Умствена изостаналост се открива при повече от половината пациенти, обикновено незначителна. Често се отбелязват поведенчески особености, дезинхибиране и разстройство с дефицит на вниманието. Речта обикновено е по-добре развита от другите интелектуални области. Степента на намаляване на интелигентността не корелира с тежестта на соматичните разстройства [Marincheva G.S., 1988]. В изолирани случаи малформации на центр нервна система(хидроцефалия, спина бифида), тромбоемболични церебрални инфаркти, вероятно свързани със съдова хипоплазия.
Пороци вътрешни органисъс синдрома на Noonan са доста характерни. Най-типичните сърдечно-съдови аномалии са: клапна стенозабелодробна артерия (около 60% от пациентите), хипертрофична кардиомиопатия(20–30%), структурни аномалии митрална клапа, дефекти на междупредсърдната преграда, тетралогия на Fallot; Коарктация на аортата е описана само при мъже.
При една трета от пациентите се регистрират дефекти на пикочната система (бъбречна хипоплазия, удвояване на таза, хидронефроза, мегауретер и др.).
Доста често при синдром на Noonan се наблюдава повишено кървене, особено когато хирургични интервенции V устната кухинаи назофаринкса. са открити различни дефектикоагулация: недостатъчност на тромбоцитната система, понижени нива на коагулационните фактори, особено XI и XII, увеличено тромбопластиново време. Има съобщения за комбинация от синдром на Noonan с левкемия и рабдомиосаркома, което може да показва леко повишаване на риска от злокачествено заболяване при тези пациенти.
Таблица 1 представя характеристиките на фенотипа при синдрома на Noonan, които се променят с възрастта на пациента. Таблица 2 показва корелацията между фенотипа и генотипа при синдрома на Noonan.
маса 1. Типични черти на лицето на пациенти със синдром на Noonan по възраст
| Чело, лице, коса | очи | Уши | нос | Устата | Шия | |
| новородено* | Високо чело, ниска линия на косата в задната част на главата | Хипертелоризъм, наклонен надолу палпебрални фисури, гънка от епикантус | – | Къс и широк вдлъбнат корен, обърнат нагоре връх | Дълбоко вдлъбнат филтрум, високи широки върхове на червената граница на устните, микрогнатия | Излишна кожа на гърба на главата |
| Бебе (2–12 месеца) | Голяма глава, високо и изпъкнало чело | Хипертелоризъм, птоза или дебели увиснали клепачи | – | Къс и широк вдлъбнат корен | – | – |
| Дете (1–12 години) | Груби черти, дълго лице | – | – | – | – | – |
| Тийнейджъри (12–18 години) | Миопатично лице | – | – | Мостът е висок и тънък | – | Явно образуване на цервикални гънки |
| Възрастен (>18 години) | Отличителните черти на лицето са изтънчени, кожата изглежда тънка и прозрачна | – | – | Изпъкнала назолабиална гънка | – | – |
| Всички възрасти | – | Сини и зелени ириси, ромбовидни вежди | Ниски, завъртяни назад уши с плътни гънки | – | – | – |
таблица 2. Корелации между генотип и фенотип при синдром на Noonan*
| Сърдечно-съдовата система | Височина | развитие | Кожа и коса | други | |
| PTPN11 (приблизително 50%) | Стенозата на белодробния ствол е по-изразена; по-малко – хипертрофична кардиомиопатия и дефект на предсърдната преграда | По-къса височина; по-ниска концентрация на IGF1 | Пациентите с N308D и N308S имат лек спадили нормална интелигентност | – | По-изразено хеморагична диатезаи ювенилна миеломоноцитна левкемия |
| SOS1 (приблизително 10%) | По-малък дефект на предсърдната преграда | По-висок растеж | По-малко намаляване на интелигентността, забавено развитие на речта | Подобно на сърдечно-кожно-лицевия синдром | – |
| RAF1 (приблизително 10%) | По-тежка хипертрофична кардиомиопатия | – | – | | Повече ▼ родилни петна, лентиго, петна от кафе с мляко | – |
| КРАС (<2%) | – | – | По-тежко когнитивно забавяне | Подобно на сърдечно-кожно-лицевия синдром | – |
| NRAS (<1%) | – | – | – | – | – |
Данни от лабораторни и функционални изследвания
Няма специфични биохимични маркери за диагностициране на синдрома на Нунан. При някои пациенти се установява намаление на спонтанната нощна секреция на растежен хормон при нормален отговор на фармакологични стимулиращи тестове (клонидин и аргинин), понижение на нивото на соматомедин-С и намаляване на отговора на соматомедините към приложението на хормон на растежа.
Критерии за диагностика
Диагнозата на синдрома на Нунан се поставя въз основа на клинични признаци, в някои случаи диагнозата се потвърждава от резултатите от молекулярно-генетично изследване. Критериите за диагностициране на синдрома включват наличието на характерно лице (с нормален кариотип) в комбинация с един от следните признаци: сърдечна патология, нисък ръст или крипторхизъм (при момчета), забавен пубертет (при момичета). За да се идентифицира сърдечно-съдовата патология, е необходимо да се проведе ултразвуково изследване на сърцето с динамично определяне на размера на кухините и стената на вентрикулите. Пренаталната диагностика на заболяването е възможна с помощта на ултразвуково наблюдение, което позволява идентифициране на сърдечни дефекти и аномалии в структурата на шията.
Диференциална диагноза
При момичетата диференциалната диагноза е предимно синдром на Turner; Цитогенетичното изследване може да изясни диагнозата. Фенотипните признаци на синдрома на Нунан се откриват при редица други заболявания: синдром на Уилямс, синдром на LEOPARD, синдром на Дубовиц, сърдечно-лицево-кожен синдром, Cornelia de Lange, Cohen, Rubinstein-Taybi и др. Точното идентифициране на тези заболявания ще бъде възможно само чрез провеждане на молекулярно генетични изследвания на всеки синдром със значителен клиничен материал, който в момента се развива активно.
Лечение
Лечението на пациенти със синдром на Noonan е насочено към премахване на дефекти на сърдечно-съдовата система, нормализиране на умствените функции, стимулиране на растежа и сексуалното развитие. За лечение на пациенти с дисплазия на белодробната клапа, наред с други методи, успешно се използва балонна валвулопластика. За стимулиране на умственото развитие се използват ноотропни и съдови лекарства. Лекарствата, насочени към стимулиране на сексуалното развитие, са показани главно при пациенти с крипторхизъм. Препаратите с човешки хорион гонадотропин се използват в дозировки, специфични за възрастта. В по-напреднала възраст - при наличие на хипогонадизъм - тестостеронови препарати. През последните години рекомбинантни форми на човешки растежен хормон се използват при лечението на пациенти със синдром на Noonan. Клиничните данни се потвърждават от повишаване на нивото на соматомедин-С и специфичния свързващ протеин по време на терапията. Крайният ръст на пациентите, получаващи терапия с растежен хормон за дълго време, в някои случаи надвишава средния ръст на членовете на семейството.
Прогноза за цял живот се определя от тежестта на сърдечно-съдовата патология.
Предотвратяване заболяване се основава на данни от медицинско генетично консултиране.
Медицинско генетично консултиране
При провеждане на медицинско генетично консултиране трябва да се изхожда от автозомно-доминантния тип наследяване и високия (50%) риск от повторение на заболяването в семейството с наследствени форми. За да се идентифицира естеството на вида на наследството, е необходимо да се извърши задълбочен преглед на родителите, тъй като синдромът може да се прояви с минимални клинични симптоми. В момента е разработена и се усъвършенства молекулярно-генетична диагностика на заболяването чрез типизиране на мутации в гените: PTPN11, SOS1, RAF1, KRAS, NRAS и др. Разработват се методи за пренатална диагностика на заболяването.
Клинично наблюдение
Момче Г., 9 години (снимка 3), е наблюдавано по местоживеене от генетик с диагноза „хромозомна патология?, синдром на Уилямс (особен фенотип, удебеляване на платната на митралната клапа, хиперкалциемия веднъж на всеки 3 години) ?.

. Характеристики на фенотипа на дете със синдром на Noonan (удължен лицев скелет с „пухкави бузи“, къс врат, криловидни гънки на шията, скъсен нос с отворени напред ноздри, пухкави устни, наклонена брадичка, антимонголоиден разрез на палпебралните фисури , неправилно захапване, макростомия)
Оплаквания за намалена памет, умора, намалени темпове на растеж.
Семейна история : родителите са руски по националност, нямат кръвна връзка и нямат професионални рискове, здрави. Височината на бащата е 192 см, височината на майката е 172 см. В родословието няма случаи на психични заболявания, епилепсия или изоставане в развитието.
История на живота и болестта : момче от 2-ра бременност (1-ва бременност - m/a), протекла през цялото време със заплаха от спонтанен аборт, придружена от полихидрамнион. Първо раждане, навреме, бързо, тегло при раждане – 3400 гр., дължина – 50 см. Изпищя веднага, оценка по Апгар – 7/9 точки. При раждането неонатологът обърна внимание на необичайния фенотип на детето и препоръча изследване на кариотипа, резултатът беше 46, XY (нормален мъжки кариотип). Има съмнение за вроден хипотиреоидизъм, изследван е профил на щитовидната жлеза и резултатът е нормален тиреоиден статус. След това детето е наблюдавано от генетик с предполагаема диагноза синдром на Уилямс. Ранният постнатален период е без особености. Моторно развитие по възраст, първи думи - до една година, фразова реч - на 2 години 3 месеца.
На 8-годишна възраст е консултиран от ендокринолог относно намален растеж, умора и намалена памет. Рентгеновото изследване на ръцете показва умерено изоставане в костната възраст (BA) от паспортната възраст (BA съответства на 6 години). Проучване на профила на щитовидната жлеза разкрива умерено увеличение на тиреостимулиращия хормон при нормални нива на свободен Т4 и други показатели; Ехография на щитовидна жлеза - без патология. Предписана е хормонална терапия с последващо динамично наблюдение.
Като се има предвид несигурността на диагнозата по местоживеене, генетикът изпрати детето в Московския регионален консултативен и диагностичен център за деца, за да изясни диагнозата.
Данни от обективно изследване:
Височина – 126 см, тегло – 21 кг.
Физическото развитие е под средното, хармонично. Растеж Sds съответства на –1 (норма – –2+2). Характеристики на фенотипа (снимка 3): удължен лицев скелет с „пухкави бузи“, къс врат, птеригоидни гънки на шията, нисък растеж на косата на шията, скъсен нос с отворени напред ноздри, пухкави устни, наклонена брадичка, антимонголоиден разрез на палпебрални фисури, малоклузия, макростомия, хипертелоризъм на зърната, асиметрия на гръдния кош, непълна кожна синдактилия на 2-3-ти пръсти на краката, тежка хипермобилност на интерфалангеалните стави, чупливи, сухи нокти. Вътрешни органи – без особености. Сексуално развитие – Танер I (което съответства на периода преди пубертета).
Данни от лабораторни и функционални изследвания:
Клиничният анализ на кръвта и урината е нормален.
Биохимичен кръвен тест - показателите са в нормални граници.
Тиреоиден профил (TSH) – 7,5 µIU/ml (норма – 0,4–4,0), останалите показатели са в норма.
Соматотропен хормон (GH) – 7 ng/ml (норма – 7–10), соматомедин-С – 250 ng/ml (норма – 88–360).
Ехография на щитовидна жлеза - без патология.
Ехография на вътрешни органи - без особености.
ЕКГ – синусова тахикардия, нормално положение на електрическата ос на сърцето.
ЕхоКГ - I степен MVP с минимална регургитация, миксоматозно удебеляване на платната на митралната клапа, допълнителна хорда в кухината на лявата камера.
R-графия на гръбначния стълб – дясностранна сколиоза на торакален гръбначен стълб I степен.
R-графия на ръцете с хват на предмишниците - костна възраст 7–8 години.
Не са регистрирани ЕЕГ модели на епилептична активност.
ЯМР на мозъка - без патологични изменения.
Аудиограма – без патология.
ДНК диагностика: молекулярно-генетично изследване - не са открити делеции на изследваните локуси на критичния участък на хромозома 7; мутация Gly434Ary (1230G>A) е открита в 11-ия екзон на гена SOS1 (анализ на гена PTPN11 - не са открити мутации), което е характерно за синдрома на Noonan.
Специализирани консултации:
Ендокринолог– субклиничен хипотиреоидизъм, непълна медикаментозна компенсация.
Окулист– астигматизъм.
Невролог– вегетативно-съдова дистония. Невротични реакции.
Кардиолог– функционална кардиопатия.
Хирург-ортопед- лоша поза. Деформация на гръдния кош.
Генетик– Синдром на Нунан.
Като се вземат предвид фенотипа на детето, медицинската история и резултатите от допълнителни изследвания, беше поставена диагноза синдром на Noonan, която беше потвърдена от резултата от молекулярно-генетично изследване.
По този начин представеното клинично наблюдение показва трудностите на диференциално диагностичното търсене, необходимостта от интегриране на отделни признаци в общия фенотип на конкретно патологично състояние за целенасочена навременна диагностика на отделни форми на наследствени заболявания и значението на молекулярно-генетичните методи за изясняване диагнозата. Навременната диагноза и изясняването на генезиса на всеки синдром са особено важни, тъй като те позволяват да се намери оптимален подход към лечението на тези състояния и предотвратяване на възможни усложнения (до и включително увреждане на детето); предотвратяване на повторна поява на наследствени заболявания в засегнатите семейства (медико-генетично консултиране). Това налага необходимостта лекарите от различни специалности ясно да се ориентират в потока на наследствено обусловената патология.
Библиография:
- Baird P., Синдром на De Jong B. Noonan (XX и XY фенотип на Turner) в три поколения на семейство // J. Pediatr., 1972, vol. 80, стр. 110–114.
- Хасегава Т., Огата Т. и др. Коарктация на аортата и бъбречна хупоплазия при момче с повърхностни аномалии на Turner/Noonan и кариотип 46, XY: клиничен модел за възможно увреждане на предполагаем лимфогенен ген(и) за соматични стигмати на Turner // Hum. Генет., 1996, кн. 97, r. 564–567.
- Федотова Т.В., Кадникова В.А. и др. Клиничен и молекулярно-генетичен анализ на синдрома на Noonan. Материали от VI конгрес на Руското дружество по медицинска генетика. Медицинска генетика, приложение към бр.5, 2010, с.184.
- Ward K.A., Moss C., McKeown C. Кардио-фацио-кутанен синдром: проява на синдрома на Noonan? // Br. J. Dermatol., 1994, том. 131, стр. 270–274.
- Municchi G., Pasquino A.M. и др. Лечение с хормон на растежа при синдром на Noonan: доклад за четири случая, които са достигнали крайна височина // Horm. Res., 1995, том. 44, r. 164–167.
ГЛАВА 5 ИЗМЕНЧИВОСТ НА ОРГАНИЗМА
ГЛАВА 5 ИЗМЕНЧИВОСТ НА ОРГАНИЗМА
Пълна информация
Променливостта на един организъм е променливостта на неговия геном, която определя генотипните и фенотипните различия на човек и причинява еволюционното разнообразие на неговите генотипове и фенотипове (вижте глави 2 и 3).
Вътрематочното развитие на ембриона, ембриона, плода, по-нататъшното постнатално развитие на човешкото тяло (детство, детство, юношество, юношество, зряла възраст, стареене и смърт) се осъществяват в съответствие с генетичната програма на онтогенезата, формирана от сливането на майчини и бащини геноми (вижте глави 2 и 12).
По време на онтогенезата геномът на тялото на индивида и информацията, кодирана в него, претърпяват непрекъснати трансформации под въздействието на факторите на околната среда. Промените, които настъпват в генома, могат да се предават от поколение на поколение, причинявайки променливост в характеристиките и фенотипа на организма при потомците.
В началото на 20в. Германският зоолог W. Hacker идентифицира клон на генетиката, посветен на изследването на връзките и взаимоотношенията между генотипове и фенотипове и анализа на тяхната променливост, и го нарече феногенетика.
В момента феногенетиците разграничават два класа променливост: ненаследствена (или модификация), която не се предава от поколение на поколение, и наследствена, която се предава от поколение на поколение.
На свой ред наследствената изменчивост също се разделя на два класа: комбинирана (рекомбинация) и мутационна. Променливостта на първия клас се определя от три механизма: случайни срещи на гамети по време на оплождането; кросинговър или мейотична рекомбинация (обмен на равни участъци между хомоложни хромозоми в профазата на първото разделение на мейозата); независимо разминаване на хомоложни хромозоми към полюсите на делене по време на образуването на дъщерни клетки по време на митоза и мейоза. Вариативност на второто
клас се причинява от точкови, хромозомни и геномни мутации (виж по-долу).
Нека последователно разгледаме различните класове и видове променливост на организма на различни етапи от неговото индивидуално развитие.
Променливост по време на оплождане на гамети и началото на функционирането на генома на зараждащия се организъм
Майчиният и бащиният геном не могат да функционират отделно един от друг.
Само два родителски генома, обединени в зигота, осигуряват произхода на молекулярния живот, появата на ново качествено състояние - едно от свойствата на биологичната материя.
На фиг. Фигура 23 показва резултатите от взаимодействието на два родителски генома по време на оплождане на гамети.
Според формулата на оплождане: зигота = яйцеклетка + сперма, началото на развитието на зиготата е моментът на образуване на двойна (диплоидна), когато се срещнат два хаплоидни набора от родителски гамети. Тогава възниква молекулярният живот и се стартира верига от последователни реакции, базирани първо на експресията на гените на генотипа на зиготата, а след това на генотипите на дъщерните соматични клетки, произлезли от нея. Индивидуалните гени и групи от гени в генотипите на всички клетки на тялото започват да се „включват“ и „изключват“ по време на изпълнението на генетичната програма на онтогенезата.
Водещата роля в събитията, които се случват, принадлежи на яйцето, което има в ядрото и цитоплазмата всичко необходимо за покълването.
Ориз. 23.Резултати от взаимодействието на два родителски генома по време на оплождане на гамети (снимки съответно от www.bio.1september.ru; www.bio.fizteh.ru; www.vetfac.nsau.edu.ru)
развитие и продължаване на живота, структурните и функционални компоненти на ядрото и цитоплазмата (същността биологичен матриархат).Спермата съдържа ДНК и не съдържа цитоплазмени компоненти. Прониквайки в яйцеклетката, ДНК на спермата влиза в контакт с нейната ДНК и по този начин в зиготата се „включва“ основният молекулярен механизъм, който функционира през целия живот на организма: взаимодействието ДНК-ДНК на два родителски генома. Строго погледнато, генотипът е активиран, представен от приблизително равни части ДНК нуклеотидни последователности от майчин и бащин произход (без да се взема предвид mtDNA на цитоплазмата). Нека опростим казаното: началото на молекулярния живот в зиготата е нарушение на постоянството на вътрешната среда на яйцето (неговата хомеостаза), а целият последващ молекулен живот на многоклетъчния организъм е желанието за възстановяване на хомеостазата изложени на фактори на околната среда или баланса между две противоположни състояния: стабилност От една странаи променливост с друг.Това са причинно-следствените връзки, които определят възникването и непрекъснатостта на молекулярния живот на организма по време на онтогенезата.
Сега нека обърнем внимание на резултатите и значението на изменчивостта на генома на един организъм като продукт на еволюцията. Първо, нека разгледаме въпроса за уникалността на генотипа на зиготата или прогениторната клетка на всички клетки, тъкани, органи и системи на тялото.
Самото оплождане се случва случайно: една женска гамета се опложда само от една мъжка гамета от 200-300 милиона сперматозоиди, съдържащи се в еякулата на мъжа. Очевидно е, че всяка яйцеклетка и всеки сперматозоид се отличават един от друг по много генотипни и фенотипни характеристики: наличие на променени или непроменени гени в състава и комбинациите (резултати от комбинираната променливост), различни последователности от нуклеотидни последователности на ДНК, различни размери, форми , функционална активност (мотилитет), зрялост на гаметите и т.н. Именно тези различия ни позволяват да говорим за уникалността на генома на всяка гамета и следователно за генотипа на зиготата и целия организъм: злополуката на оплождането гамети осигурява раждането на генетично уникален индивидуален организъм.
С други думи, молекулярният живот на човека (както животът на биологичното същество като цяло) е „дар от съдбата” или, ако искате, „божествен дар”, защото вместо даден индивид със същите
имаше възможност да са родени генетично различни братя и сестри.
Сега нека продължим нашата дискусия за баланса между стабилността и променливостта на наследствения материал. В широк смисъл поддържането на такъв баланс е едновременното запазване и промяна (трансформация) на стабилността на наследствения материал под въздействието на вътрешни (хомеостаза) и външни фактори на околната среда (реакционна норма). Хомеостазата зависи от генотипа, причинен от сливането на два генома (виж фиг. 23). Скоростта на реакцията се определя от взаимодействието на генотипа с факторите на околната среда.
Норма и диапазон на реакция
Специфичният начин, по който тялото реагира в отговор на факторите на околната среда, се нарича норма на реакция.Именно гените и генотипът са отговорни за развитието и обхвата на модификациите на индивидуалните характеристики и фенотипа на целия организъм. В същото време не всички възможности на генотипа се реализират във фенотипа, т.е. фенотип е конкретен (за индивид) случай на прилагане на генотип при специфични условия на околната среда. Ето защо, например, между монозиготни близнаци, които имат напълно идентични генотипове (100% общи гени), се разкриват забележими фенотипни разлики, ако близнаците растат в различни условия на околната среда.
Нормата на реакция може да бъде тясна или широка. В първия случай стабилността на индивидуална черта (фенотип) се поддържа почти независимо от влиянията на околната среда. Примери за гени с тясна норма на реакция или непластични гениса гени, кодиращи синтеза на антигени на кръвна група, цвят на очите, къдрене на косата и др. Действието им е еднакво при всякакви (съвместими с живота) външни условия. Във втория случай стабилността на индивидуалната черта (фенотип) се променя в зависимост от влиянието на околната среда. Пример за гени с широка скорост на реакция или пластмасови гени- гени, които контролират броя на червените кръвни клетки (различни за хората, които се изкачват и слизат от планината). Друг пример за широка норма на реакция е промяната в цвета на кожата (тен), свързана с интензивността и времето на излагане на ултравиолетово лъчение върху тялото.
Говорейки за диапазон на реакция,трябва да се имат предвид фенотипните различия, които се появяват в индивида (неговия генотип) в зависимост от
„изчерпани“ или „обогатени“ условия на околната среда, в които се намира организмът. Според определението на И.И. Шмалхаузен (1946), „не се наследяват характеристиките като такива, а нормата на тяхната реакция към промените в условията на съществуване на организмите“.
По този начин нормата и обхватът на реакцията са границите на генотипната и фенотипната променливост на организма при промяна на условията на околната среда.
Трябва също да се отбележи, че сред вътрешните фактори, които влияят върху фенотипното проявление на гените и генотипа, полът и възрастта на индивида са от определено значение.
Външните и вътрешните фактори, които определят развитието на признаци и фенотипове, са включени в трите групи основни фактори, посочени в главата, включително гени и генотип, механизми на междумолекулни (ДНК-ДНК) и междугенни взаимодействия между родителските геноми и фактори на околната среда.
Разбира се, основата за адаптиране на организма към условията на околната среда (основата на онтогенезата) е неговият генотип. По-специално, индивиди с генотипове, които не потискат негативните ефекти на патологични гени и фактори на околната среда, оставят по-малко потомство от тези индивиди, при които нежеланите ефекти са потиснати.
Вероятно генотипите на по-жизнеспособни организми включват специални гени (модификаторни гени), които потискат действието на „вредните“ гени по такъв начин, че вместо това алелите от нормалния тип стават доминиращи.
НЕНАСЛЕДСТВЕНА ИЗМЕНЛИВОСТ
Говорейки за ненаследствена променливост на генетичния материал, нека отново разгледаме пример за широка норма на реакция - промяна в цвета на кожата под въздействието на ултравиолетово лъчение. “Тан” не се предава от поколение на поколение, т.е. не се предава по наследство, въпреки че в възникването му участват пластични гени.
По същия начин не се наследяват резултатите от наранявания, белези от промени в тъканите и лигавиците, дължащи се на изгаряне, измръзване, отравяне и много други признаци, причинени единствено от фактори на околната среда. В същото време трябва да се подчертае: ненаследствените промени или модификации са свързани с наследствени
естествени свойства на даден организъм, тъй като те се формират на фона на определен генотип при специфични условия на околната среда.
Наследствена комбинирана изменчивост
Както беше посочено в началото на главата, в допълнение към механизма на случайни срещи на гамети по време на оплождането, комбинираната променливост включва механизмите на кросинговър при първото делене на мейозата и независимото разминаване на хромозомите към полюсите на делене по време на образуването на дъщеря клетки по време на митоза и мейоза (виж глава 9).
Кросингоувър при първото мейотично делене
Поради механизма пресичаневръзката на гените с хромозомата редовно се нарушава в профазата на първото разделение на мейозата в резултат на смесване (обмен) на гени от бащин и майчин произход (фиг. 24).
В началото на 20в. при отваряне на прелеза над Т.Х. Морган и неговите ученици предполагат, че кръстосването между два гена може да се случи не само в една, но и в две, три (съответно двойно и тройно кръстосване) и повече точки. Потискането на пресичането беше отбелязано в райони непосредствено до пунктовете за обмен; това потискане се наричаше намеса.
В крайна сметка беше изчислено: за една мъжка мейоза има от 39 до 64 хиазми или рекомбинации, а за една женска мейоза има до 100 хиазми.
 Ориз. 24.Схема на кросинговър при първото разделение на мейозата (според Шевченко В.А. и др., 2004):
Ориз. 24.Схема на кросинговър при първото разделение на мейозата (според Шевченко В.А. и др., 2004):
а - сестрински хроматиди на хомоложни хромозоми преди началото на мейозата; b - те са по време на пахитена (видима е спирализацията им); c - те също са по време на диплотена и диакинеза (стрелките показват местата на преминаване през хиазма или области на обмен)
В резултат на това те заключиха, че връзката на гените с хромозомите непрекъснато се нарушава по време на кросингоувъра.
Фактори, влияещи върху кросингоувъра
Кросингоувърът е един от редовните генетични процеси в тялото, контролиран от много гени както директно, така и чрез физиологичното състояние на клетките по време на мейоза и дори митоза.
Факторите, влияещи върху преминаването, включват:
Хомо- и хетерогаметичен пол (говорим за митотичен кросинговърпри мъже и жени от еукариоти като дрозофила и копринена буба); По този начин при Drosophila кросинговърът протича нормално; при копринената буба то е или нормално, или липсва; при хората трябва да се обърне внимание на смесения („трети“) пол и по-специално на ролята на кросингоувъра в аномалиите на половото развитие при мъжкия и женския хермафродитизъм (виж Глава 16);
структура на хроматина; честотата на кръстосване в различни региони на хромозомите се влияе от разпределението на хетерохроматичните (перицентромерни и теломерни региони) и еухроматичните региони; по-специално, в перицентромерни и теломерни области, честотата на кръстосване е намалена и разстоянието между гените, определено от честотата на кръстосване, може да не съответства на действителното;
Функционално състояние на тялото; С напредването на възрастта степента на хромозомна спирализация и скоростта на клетъчното делене се променят;
генотип; съдържа гени, които увеличават или намаляват честотата на кръстосване; „шкафчета“ на последните са хромозомни пренареждания (инверсии и транслокации), които усложняват нормалното конюгиране на хромозомите в зиготената;
Екзогенни фактори: излагане на температура, йонизиращо лъчение и концентрирани солеви разтвори, химически мутагени, лекарства и хормони, които обикновено увеличават честотата на кросингоувъра.
Честотата на мейотичен и митотичен кросингоувър и SCO понякога се използва за оценка на мутагенния ефект на лекарства, канцерогени, антибиотици и други химични съединения.
Неравностойно пресичане
В редки случаи при кросинговър се наблюдават прекъсвания в асиметрични точки на сестрински хроматиди и те се разменят
са разделени на неравни области помежду си - това е неравномерно пресичане.
В същото време са описани случаи, когато по време на митоза се наблюдава митотична конюгация (неправилно сдвояване) на хомоложни хромозоми и възниква рекомбинация между несестрински хроматиди. Това явление се нарича генна конверсия.
Значението на този механизъм е трудно да се надценява. Например, в резултат на неправилно сдвояване на хомоложни хромозоми по страничните повторения, може да възникне удвояване (дупликация) или загуба (делеция) на хромозомния регион, съдържащ гена PMP22, което ще доведе до развитие на наследствена автозомно-доминантна моторно-сензорна невропатия на Charcot-Marie-Tooth.
Неравномерният кросингоувър е един от механизмите за възникване на мутации. Например, периферният протеин миелин е кодиран от гена PMP22, разположен на хромозома 17 и имащ дължина около 1,5 милиона bp. Този ген е ограден от две хомоложни повторения с дължина приблизително 30 kb. (повторенията са разположени по фланговете на гена).
Особено много мутации в резултат на неравномерно кръстосване се срещат в псевдогените. Тогава или фрагмент от един алел се прехвърля към друг алел, или фрагмент от псевдоген се прехвърля към ген. Например, подобна мутация се наблюдава, когато псевдогенна последователност се прехвърли към 21-хидроксилазния ген (CYP21B) при адреногенитален синдром или вродена надбъбречна хиперплазия (вижте глави 14 и 22).
В допълнение, поради рекомбинации по време на неравен кросингоувър, могат да се образуват множество алелни форми на гени, кодиращи HLA клас I антигени.
Независимо разминаване на хомоложни хромозоми към полюсите на делене по време на образуването на дъщерни клетки по време на митоза и мейоза
Поради процеса на репликация, който предшества митозата на соматична клетка, общият брой на ДНК нуклеотидните последователности се удвоява. Образуването на една двойка хомоложни хромозоми става от две бащини и две майчини хромозоми. Когато тези четири хромозоми се разпределят в две дъщерни клетки, всяка клетка ще получи една бащина и една майчина хромозома (за всяка двойка хромозомен набор), но коя от двете, първата или втората, не е известно. Възниква
произволно разпределение на хомоложни хромозоми. Лесно е да се изчисли: поради различни комбинации от 23 двойки хромозоми, общият брой на дъщерните клетки ще бъде 2 23, или повече от 8 милиона (8 χ 10 6) варианта на комбинации от хромозоми и гени, разположени върху тях. Следователно, с произволното разпределение на хромозомите в дъщерните клетки, всяка от тях ще има свой собствен уникален кариотип и генотип (собствена версия на комбинацията от хромозоми и гени, свързани с тях, съответно). Трябва да се отбележи, че има патологичен вариант на разпределението на хромозомите в дъщерните клетки. Например, влизането в една от двете дъщерни клетки само на една Х хромозома (бащин или майчин произход) ще доведе до монозомия (синдром на Шерешевски-Търнър, кариотип 45, XO), влизането на три идентични автозоми ще доведе до тризомия (Даун синдром, 47,XY,+21; Патау, 47,ХХ,+13 и Едвадса, 47,ХХ,+18;
Както беше отбелязано в Глава 5, две бащини или две майчини хромозоми на произход могат едновременно да влязат в една дъщерна клетка - това е униродителска изодизомия за специфична двойка хромозоми: синдром на Силвър-Ръсел (две майчини хромозоми 7), синдром на Беквит-Видеман (две бащини хромозоми). хромозоми 11), Angelman (две бащини хромозоми 15), Prader-Willi (две майчини хромозоми 15). Като цяло обемът на нарушенията в разпределението на хромозомите достига 1% от всички хромозомни нарушения при хората. Тези разстройства са от голямо еволюционно значение, тъй като създават популационно разнообразие на човешки кариотипове, генотипове и фенотипове. Освен това всеки патологичен вариант е уникален продукт на еволюцията.
В резултат на второто мейотично делене се образуват 4 дъщерни клетки. Всеки от тях ще получи една майчина или бащина хромозома от всичките 23 хромозоми.
За да избегнем възможни грешки в по-нататъшните си изчисления, ще приемем за правило: в резултат на второто мейотично делене се образуват също 8 милиона варианта на мъжки гамети и 8 милиона варианта на женски гамети. Тогава отговорът на въпроса какъв е общият обем на вариантните комбинации от хромозоми и гени, разположени върху тях, когато се срещнат две гамети, е следният: 2 46 или 64 χ 10 12, т.е. 64 трилиона.
Образуването на такъв (теоретично възможен) брой генотипове при среща на две гамети ясно обяснява значението на хетерогенността на генотипите.
Стойността на комбинираната променливост
Комбинативната изменчивост е важна не само за хетерогенността и уникалността на наследствения материал, но и за възстановяването (поправянето) на стабилността на молекулата на ДНК, когато и двете вериги са увредени. Пример е образуването на едноверижна ДНК празнина срещу непоправена лезия. Получената празнина не може да бъде точно коригирана без включването на нормалната ДНК верига в ремонта.
Мутационна изменчивост
Наред с уникалността и хетерогенността на генотиповете и фенотипите в резултат на комбинираната вариабилност, огромен принос за вариабилността на човешкия геном и феномен има наследствената мутационна вариабилност и произтичащата от това генетична хетерогенност.
Вариациите в ДНК нуклеотидните последователности могат чисто условно да се разделят на мутации и генетичен полиморфизъм (виж Глава 2). В същото време, ако хетерогенността на генотипите е постоянна (нормална) характеристика на променливостта на генома, тогава мутационна изменчивост- това е, като правило, неговата патология.
Патологичната вариабилност на генома се поддържа, например, от неравномерно кръстосване, неправилна дивергенция на хромозомите към полюсите на делене по време на образуването на дъщерни клетки, наличието на генетични съединения и алелни серии. С други думи, наследствената комбинирана и мутационна изменчивост се проявява при хората чрез значително генотипно и фенотипно разнообразие.
Нека изясним терминологията и разгледаме общите въпроси на теорията на мутациите.
ОБЩИ ВЪПРОСИ НА ТЕОРИЯТА НА МУТАЦИИТЕ
Мутациянастъпва промяна в структурната организация, количеството и/или функционирането на наследствения материал и синтезираните от него протеини. Тази концепция е предложена за първи път от Hugo de Vries
през 1901-1903г в работата си „Теория на мутациите“, където описва основните свойства на мутациите. Те:
Появяват се внезапно;
Предава се от поколение на поколение;
Унаследява се по доминантен тип (проявява се при хетерозиготи и хомозиготи) и рецесивен тип (проявява се при хомозиготи);
Те нямат посока („мутират“ всеки локус, причинявайки незначителни промени или засягащи жизнените показатели);
Според фенотипното си проявление те могат да бъдат вредни (повечето мутации), полезни (изключително редки) или безразлични;
Срещат се в соматични и зародишни клетки.
Освен това едни и същи мутации могат да се появят многократно.
Процес на мутацияили мутагенеза, е непрекъснато протичащ процес на образуване на мутации под въздействието на мутагени - фактори на околната среда, които увреждат наследствения материал.
Първо теория на непрекъснатата мутагенезапредложен през 1889 г. от руския учен от Санкт Петербургския университет S.I. Коржински в книгата си „Хетерогенезис и еволюция“.
Както се смята в момента, мутациите могат да се появят спонтанно, без видими външни причини, но под влияние на вътрешни условия в клетката и тялото – това са спонтанни мутации или спонтанна мутагенеза.
Мутациите, причинени изкуствено от излагане на външни фактори от физическо, химическо или биологично естество, са индуцирани мутации, или индуцирана мутагенеза.
Най-честите мутации се наричат големи мутации(например мутации в гените на мускулна дистрофия на Дюшен-Бекер, кистозна фиброза, сърповидно-клетъчна анемия, фенилкетонурия и др.). Вече са създадени търговски комплекти, които позволяват автоматичното идентифициране на най-важните от тях.
Нововъзникналите мутации се наричат нови мутации или мутации de novo.Например, те включват мутации, които са в основата на редица автозомно-доминантни заболявания, като ахондроплазия (10% от случаите на заболяването са фамилни форми), неврофиброматоза на Реклингхаузен тип I (50-70% са фамилни форми), болест на Алцхаймер, хорея на Хънтингтън .
Наричат се мутации от нормалното състояние на ген (белег) до патологично състояние прав.
Мутациите от патологично състояние на ген (белег) към нормално състояние се наричат обратни или реверсии.
Способността за връщане е установена за първи път през 1935 г. от N.V. Тимофеев-Ресовски.
Последващите мутации в гена, които потискат първичния мутантен фенотип, се наричат супресор.Потискането може да бъде интрагенен(възстановява функционалната активност на протеина; аминокиселината не отговаря на оригиналната, т.е. няма истинска обратимост) и екстрагенен(структурата на tRNA се променя, в резултат на което мутантната tRNA включва друга аминокиселина в полипептида вместо тази, кодирана от дефектния триплет).
Мутациите в соматичните клетки се наричат соматични мутации.Те образуват патологични клетъчни клонове (набор от патологични клетки) и в случай на едновременно присъствие на нормални и патологични клетки в тялото водят до клетъчен мозаицизъм (например при наследствената остеодистрофия на Олбрайт, изразеността на заболяването зависи от броя на анормалните клетки).
Соматичните мутации могат да бъдат както фамилни, така и спорадични (нефамилни). Те са в основата на развитието на злокачествени новообразувания и процеси на преждевременно стареене.
Преди това се смяташе за аксиома, че соматичните мутации не се предават по наследство. През последните години е доказано предаването от поколение на поколение на наследствена предразположеност на 90% от мултифакторните форми и 10% от моногенните форми на рак, проявяващи се чрез мутации в соматичните клетки.
Мутациите в зародишните клетки се наричат зародишни мутации.Смята се, че те са по-редки от соматичните мутации, лежат в основата на всички наследствени и някои вродени заболявания, предават се от поколение на поколение и също могат да бъдат фамилни или спорадични. Най-изследваната област на общата мутагенеза е физическата и по-специално, радиационна мутагенеза.Всички източници на йонизиращо лъчение са вредни за човешкото здраве, като правило имат мощен мутагенен, тератогенен и канцерогенен ефект. Мутагенният ефект на единична доза радиация е много по-висок от този на хроничното облъчване; Доза радиация от 10 rad удвоява скоростта на мутация при хората. Доказано е, че йонизиращото лъчение може да предизвика мутации, които водят до
към наследствени (вродени) и онкологични заболявания, а ултравиолетовите - за индуциране на грешки в репликацията на ДНК.
Най-голямата опасност е химическа мутагенеза.В света има около 7 милиона химични съединения. Приблизително 50-60 хиляди химически вещества се използват постоянно в националната икономика, в производството и в ежедневието. Около хиляда нови съединения се въвеждат в практиката всяка година. От тях 10% са в състояние да предизвикат мутации. Те включват хербициди и пестициди (делът на мутагените сред тях достига 50%), както и редица лекарства (някои антибиотици, синтетични хормони, цитостатици и др.).
Има и биологична мутагенеза.Биологичните мутагени включват: чужди протеини на ваксини и серуми, вируси (варицела, морбили рубеола, полиомиелит, херпес симплекс, СПИН, енцефалит) и ДНК, екзогенни фактори (лошо протеиново хранене), хистаминови съединения и техните производни, стероидни хормони (ендогенни фактори) . Укрепване на ефекта на външните мутагени комутагени(токсини).
Историята на генетиката има много примери за важността на връзките между гените и чертите. Една от тях е класификацията на мутациите в зависимост от техния фенотипен ефект.
Класификация на мутациите в зависимост от техния фенотипен ефект
Тази класификация на мутациите е предложена за първи път през 1932 г. от G. Möller. Според класификацията са идентифицирани следните:
Аморфни мутации. Това е състояние, при което белегът, контролиран от патологичния алел, не се изразява, тъй като патологичният алел е неактивен в сравнение с нормалния алел. Такива мутации включват гена за албинизъм (11q14.1) и около 3000 автозомно-рецесивни заболявания;
Антиморфни мутации. В този случай стойността на признака, контролиран от патологичния алел, е противоположна на стойността на признака, контролиран от нормалния алел. Такива мутации включват гени на около 5-6 хиляди автозомно-доминантни заболявания;
Хиперморфни мутации. В случай на такава мутация, белегът, контролиран от патологичния алел, е по-изразен от белегът, контролиран от нормалния алел. Пример - gete-
розиготни носители на гени за заболявания на нестабилност на генома (виж глава 10). Техният брой е около 3% от населението на Земята (почти 195 милиона души), а броят на самите заболявания достига 100 нозологии. Сред тези заболявания: анемия на Фанкони, атаксия телеангиектазия, пигментна ксеродерма, синдром на Блум, прогероидни синдроми, много форми на рак и др. Освен това честотата на рак при хетерозиготни носители на гените за тези заболявания е 3-5 пъти по-висока от нормалната, и при самите пациенти (хомозиготи за тези гени), честотата на рак е десетки пъти по-висока от нормалното.
Хипоморфни мутации. Това е състояние, при което експресията на черта, контролирана от патологичен алел, е отслабена в сравнение с чертата, контролирана от нормален алел. Такива мутации включват мутации в гените за синтез на пигмент (1q31; 6p21.2; 7p15-q13; 8q12.1; 17p13.3; 17q25; 19q13; Xp21.2; Xp21.3; Xp22), както и повече от 3000 форми на автозомно-рецесивни заболявания.
Неоморфни мутации. Твърди се, че такава мутация възниква, когато чертата, контролирана от патологичния алел, е с различно (ново) качество в сравнение с чертата, контролирана от нормалния алел. Пример: синтез на нови имуноглобулини в отговор на проникването на чужди антигени в тялото.
Говорейки за трайното значение на класификацията на G. Möller, трябва да се отбележи, че 60 години след нейното публикуване фенотипните ефекти на точковите мутации са разделени на различни класове в зависимост от ефекта, който имат върху структурата на протеиновия продукт на гена и /или нивото му на изразяване.
По-специално, Нобеловият лауреат Виктор Маккузик (1992) идентифицира мутации, които променят последователността на аминокиселините в протеина. Оказа се, че те са отговорни за проявата на 50-60% от случаите на моногенни заболявания, а останалите мутации (40-50% от случаите) представляват мутации, засягащи генната експресия.
Промяната в аминокиселинния състав на протеина се проявява в патологичен фенотип, например в случаи на метхемоглобинемия или сърповидно-клетъчна анемия, причинени от мутации на бетаглобиновия ген. На свой ред са изолирани мутации, засягащи нормалната генна експресия. Те водят до промяна в количеството на генния продукт и се проявяват чрез фенотипове, свързани с дефицита на определен протеин, напр.
в случаите хемолитична анемия,причинени от мутации на гени, локализирани върху автозоми: 9q34.3 (дефицит на аденилат киназа); 12p13.1 (дефицит на триозофосфат изомераза); 21q22.2 (дефицит на фосфофруктокиназа).
Класификацията на мутациите от V. McKusick (1992) е, разбира се, ново поколение класификации. В същото време, в навечерието на публикуването му, класификацията на мутациите в зависимост от нивото на организация на наследствения материал стана широко приета.
Класификация на мутациите в зависимост от нивото на организация на наследствения материал
Класификацията включва следното.
Точкови мутации(нарушаване на генната структура в различни точки).
Строго погледнато, точковите мутации включват промени в нуклеотидите (базите) на един ген, водещи до промяна в количеството и качеството на протеиновите продукти, които синтезират. Основните промени са техните замествания, вмъквания, движения или делеции, които могат да се обяснят с мутации в регулаторните региони на гените (промотор, сайт на полиаденилиране), както и в кодиращите и некодиращите региони на гените (екзони и интрони, сплайсинг) сайтове). Базовите замествания водят до три вида мутантни кодони: миссенс мутации, неутрални мутации и безсмислени мутации.
Точковите мутации се наследяват като прости менделски черти. Те са чести: 1 случай на 200-2000 раждания - първична хемохроматоза, неполипозен рак на дебелото черво, синдром на Мартин-Бел и кистозна фиброза.
Точковите мутации, които са изключително редки (1:1 500 000), са тежка комбинирана имунна недостатъчност (SCID) в резултат на дефицит на аденозин деаминаза. Понякога точковите мутации се образуват не поради излагане на мутагени, а като грешки в репликацията на ДНК. Освен това тяхната честота не надвишава 1:10 5 -1:10 10, тъй като те се коригират с помощта на системи за възстановяване на клетките с почти
Структурни мутацииили хромозомни аберации (нарушават структурата на хромозомите и водят до образуването на нови групи за генно свързване). Това са делеции (загуби), дупликации (удвоявания), транслокации (движения), инверсии (завъртане на 180°) или инсерции (вмъквания) на наследствен материал. Такива мутации са характерни за соматичните
логически клетки (включително стволови клетки). Честотата им е 1 на 1700 клетъчни деления.
Има редица синдроми, причинени от структурни мутации. Най-известните примери: синдром на "котешки плач" (кариотип: 46,ХХ,5р-), синдром на Wolf-Hirschhorn (46,ХХ,4р-), транслокационна форма на синдрома на Даун (кариотип: 47, ХУ, t ( 14;21) ).
Друг пример е левкемията. Когато се появят, генната експресия се нарушава в резултат на така наречената сепарация (транслокация между структурната част на гена и неговия промоторен участък) и следователно се нарушава протеиновият синтез.
Геномни(числово) мутации- нарушение на броя на хромозомите или техните части (водят до появата на нови геноми или техни части чрез добавяне или загуба на цели хромозоми или техни части). Произходът на тези мутации се дължи на неразделяне на хромозомите при митоза или мейоза.
В първия случай това са анеуплоиди, тетраплоиди с неразделена цитоплазма, полиплоиди с 6, 8, 10 двойки хромозоми или повече.
Във втория случай това е неразделянето на сдвоени хромозоми, участващи в образуването на гамети (монозомия, тризомия) или оплождането на едно яйце от два сперматозоида (диспермия или триплоиден ембрион).
Типичните им примери вече са цитирани повече от веднъж - това са синдромът на Шерешевски-Търнър (45, XX), синдромът на Klinefelter (47, XXY), редовната тризомия при синдрома на Даун (47, XX, +21).
във F2. Индивидите с доминантна черта могат да бъдат или хомозиготни (AA),
и хетерозиготни (Аа) носители на доминантния алел. За да се установи, е необходимо да се извърши аналитично кръстосване на такъв индивид с рецесивен хомозигот. Ако изследваният индивид е доминиращ хомозигот, тогава всички потомци от този кръст ще имат доминантна черта и в същото време ще бъдат хетерозиготи (Aa). Във втория случай, потомството ще съдържа еднакво вероятно индивиди с доминираща (Aa) и рецесивна (aa) черта.
Вече казахме, че методите на хибридологичния анализ не са приложими за хората. Определянето на вида на наследството може да се направи само въз основа на анализ на родословия. В някои случаи автозомно
доминантното заболяване е налице при един от родителите на пациента. Освен това, независимо от пола, вероятността за проявление на чертата в потомството на хетерозиготен носител на доминантна мутация е 50% и
хомозиготни – 100%. Но най-често (до 90% от случаите) доминантните заболявания са резултат от de novo мутация. В този случай те се проявяват като спорадични заболявания.
При автозомно-рецесивен тип наследяване, признакът ще отсъства при хибридите от първо поколение, но при F2 вероятността за раждане на индивиди с рецесивен признак ще бъде 25%, независимо от техния пол.
При провеждане на анализиращо кръстосване ще се наблюдава рецесивен признак, както и доминантен, в половината от потомците. Индивидите с рецесивен белег са хомозиготни носители на рецесивния алел (аа). Най-често те се появяват в потомството на хетерозиготни родители, които сами нямат рецесивен белег, но са хетерозиготни носители на мутацията. Такива родители се наричат
облигатни хетерозиготи. Според закона на Мендел вероятността да имате болно дете при облигатни хетерозиготи е 25%. Ако говорим за автозомно рецесивно заболяване, тогава родителите на засегнатото дете обикновено са здрави, но може да имат няколко засегнати деца. Деца с
Автозомно рецесивно заболяване често се ражда в кръвно-родствени бракове и вероятността да имате засегнато дете се увеличава със степента на родство между родителите. Автозомно-рецесивните мутации могат да се натрупат в популация, тъй като хетерозиготните носители не са подложени на селективен натиск. Ако родителите на болно дете не са свързани, тогава най-често те носят различни мутации в един и същ ген и
техните засегнати деца наследяват всяка от тези мутации, т.е
сложни хетерозиготи. Автозомно-рецесивен тип наследство е характерен за повечето наследствени ензимопатии.
Особености на наследяване на признаци, определени от гени
локализирани в половите хромозоми се обясняват с факта, че в Y-
има малко гени на хромозомата и практически няма хомолози на гените на Х хромозомата.
В резултат на това при мъжете се появяват рецесивни алели на повечето гени на X хромозомата. Това състояние на рецесивен алел,
когато му липсва хомолог - (a/-), се нарича хемизиготен.
Обърнете внимание, че този термин се отнася не само до гени, локализирани в половите хромозоми, но и до автозомни гени в случаите, когато регионът на локализация на този ген в една от хомоложните хромозоми е изтрит, т.е. липсва.
При Х-свързаното наследяване ще се наблюдават фенотипни разлики в потомството в зависимост от посоката на кръстосване, тоест в зависимост от наличието на черта в майката или бащата в родителското поколение. Ако чертата е доминираща и присъства в хомозиготната майка,
тогава при F1 всички индивиди, независимо от пола, ще притежават тази черта, а при F2 ще се наблюдава разделяне 3:1, като чертата липсва само при половината от мъжките индивиди. В потомството на хетерозиготна майка вероятността за раждане на индивиди с доминантна черта ще бъде 50%
независимо от пола. Ако доминиращата черта в родителското поколение е в бащата, то в първото поколение тази черта ще бъде
присъства само при дъщерите, а във втория - и при дъщерите, и при синовете с вероятност 50%.
При рецесивен тип наследяване, свързан с пола, чертата най-често ще се открива при мъжки индивиди и ще се наблюдава предаване на чертата от „дядо“ на „внук“. Никога няма да има предаване на болестта от баща на син, тъй като синът не наследява Х-хромозомата на бащата, тя винаги е от майчин произход. В повечето случаи мъжете с рецесивен белег, свързан с пола, имат 50% вероятност да се появят в потомството на хетерозиготни майки, които нямат този белег. Всички потомци на първото поколение на баща с рецесивен белег няма да притежават този белег, но половината от дъщерите му ще носят мутацията в хетерозиготно състояние и вероятността да раждат мъже с рецесивен белег, както казахме по-рано , ще бъде 50%. Типът на наследяване на признаци, определени от гените на Y хромозомата, се нарича холандричен и се характеризира с
предаване на черта от баща на син.
През последните десетилетия се натрупаха много факти, сочещи
за наличието на голям брой отклонения от Менделееви типове
наследяване. Към неменделските заболявания с нетрадиционен тип
наследството включва митохондриални заболявания, униродителски дисомии и геномни импринтинг заболявания, както и болести на разширяване,
причинени от наличието на динамични мутации. Митохондриалният или цитоплазмен тип наследство се нарича майчино.
Мъжките зародишни клетки, въпреки че съдържат много малък брой митохондрии, които осигуряват тяхната подвижност, не ги предават на своето потомство. Следователно всички митохондрии на плода, независимо от неговия пол, имат
момчета и момичета еднакво. В бъдеще ще обсъдим по-подробно всички видове наследство, използвайки примера на различни човешки заболявания.
Нека подчертаем още веднъж, че моделите на унаследяване, обсъдени по-горе, са валидни за моногенни черти. В каталога на човешките гени и генетични заболявания, който през последните няколко десетилетия е съставен с прякото участие и ръководството на изключителния медицински генетик на нашето време, Виктор А. Маккузик (McKusick V. A. Mendelian inheritance in man: a catalogue of human гени и генетично разстройство http://www.ncbi.nlm.nih.gov/, е представено описание на повече от 16 000 гена, отговорни за фенотипни черти. Начинът на наследяване е установен за приблизително 11 000 от тях и са картографирани повече от 8 000 човешки гена. Близо до
4500 гена са свързани с различни моногенни заболявания. За приблизително 4000 моногенни заболявания е установен начинът на унаследяване. Броят на автозомните заболявания надхвърля 3500, а броят на доминантните и рецесивните заболявания е приблизително еднакъв, въпреки че все още има малко повече доминиращи. Повече от 300 заболявания се наследяват по Х-свързан начин,
само няколко (не повече от 10) - Y-свързани и малко повече от 20
заболяванията се причиняват от мутации в митохондриалните гени.
В някои случаи нито един от родителите не е носител на мутацията, присъстваща в детето им. Вече писахме, че мутации в определен ген могат да възникнат de novo по време на гаметогенезата в една от родителските зародишни клетки. Някои автозомно-доминантни заболявания се дължат изцяло на de novo мутации. Те включват ахондроплазия, при която повечето пациенти имат специфична мутация в гена на рецептора на фибробластния растежен фактор 3 (Fgf). Почти всички случаи възникват de novo
заместване на една от хомоложните аминокиселини (пролин) в гените на три Fgf-
рецептори, идентифицирани при пациенти с различни наследствени форми на краниосиностоза (неправилно сливане на шевовете на черепа в
дете). Честотите на тези мутации са с три порядъка по-високи от нормалните. Местата на възникване на тези специфични мутации са сред най-променливите в човешкия геном или се смята, че са
„горещи точки” на мутагенезата и причините за тази изключително специфична нестабилност все още не са известни.
Честотите на мутациите в гените на мускулна дистрофия на Дюшен и хемофилия А са повишени. Почти 40-45% от пациентите с тези Х-свързани заболявания имат de novo мутации. Когато се предоставя медицинско генетично консултиране на такива пациенти, е много важно да се определи дали пациентът е наследил мутацията от своята хетерозиготна майка или тя е възникнала de novo.
В първия случай, в такова семейство с многократна бременност, е необходимо да се извършат определени превантивни мерки, насочени към предотвратяване на раждането на болно дете. Във втория случай рискът от повторно раждане на болно дете в дадено семейство не надвишава стойността на общата популация и това семейство не се нуждае от превантивни мерки. Ще обсъдим тази ситуация по-подробно по-късно.
Повече от един ген може да участва в генетичния контрол на по-голямата част от чертите в един организъм. В този случай те говорят за
полигенно наследство. Понякога броят на тези гени може да достигне десетки и дори стотици. Полигенното наследство е типично, по-специално,
за количествени признаци, чиито показатели могат да бъдат измерени, като ръст, тегло, продължителност на живота и много продуктивни свойства на селскостопанските растения и животни. Изменчивостта във фенотипните прояви на такива белези в популациите отговаря на нормално разпределение – Фиг. 12.
Фигура 12. Пример за нормално разпределение
Класът на полигенно унаследените признаци включва много широко разпространени човешки заболявания - атеросклероза, хипертония, захарен диабет, язвена болест, бронхиална астма и много други мултифакторни заболявания. За изследване на унаследяването на количествени и други полигенни признаци се използват статистически методи, разработени през първата половина на миналия век от Фишер.
Глава 1.6. Популационна генетика
Всеки тип организъм се характеризира с определено ниво на генотипна изменчивост, чийто характер е различен в различните популации. Изследването на генетичното разнообразие на популациите и моделите на неговото поддържане са предмет на популационната наука
генетика. Основата за развитието на тази област на генетиката е работата на С. С. Четвериков „За някои аспекти на еволюционния процес от гледна точка на съвременната генетика“, публикувана през 1926 г. Той обсъжда за първи път въпросите за поддържане на мутации в естествените популации,
влияние върху този процес на селекция и изолация, както и тяхното значение в еволюцията.
В големи популации, в които няма предпочитание при формирането на семейни двойки въз основа на свързани, национални, религиозни, социални или други характеристики (такива популации се наричат панмиктични от думата panmixia - произволно кръстосване), връзката между честотите на алелите и генотипове съответства на закона на Харди-Вайнберг, открит независимо от тези двама учени през 1908 г. За моногенните черти това звучи така: ако честотите на алелите A ia са равни на p и q, тогава честотите на хомозиготите AA iaa ще бъдат равни на p2 и q2, а хетерозиготите Aa - 2pq
съответно.
При селекция, насочена срещу определен генотипен клас, мутации или инбридинг, който се среща в близкородствени бракове и в малки географски изолирани популации, т.нар. генетични изолати, тези съотношения
са нарушени. Не само географски, но и национални, социални, религиозни и други бариери могат да доведат до генетична изолация. Мутациите, които възникват в членове на изолирани затворени общности, стават по-широко разпространени в генетичните изолати. Това явление се нарича ефект на основателя. Промяна в честотите на алелите през поредица от поколения може да възникне поради случаен подбор на индивиди, които са довели до популация или част от нея. Това явление се нарича генетичен дрейф. Ефектът на основателя е форма на генетичен дрейф. Миграцията на индивиди също може да бъде придружена от генетичен дрейф.
Инбридингът насърчава разпространението на специфични мутации, свързани с редки наследствени заболявания в генетични изолати. Честотата на някои мутации в такива популации може да се увеличи няколко, а понякога и няколко десетки пъти в сравнение с общото ниво. Класически пример, илюстриращ тези точки, е етническата група евреи от източноевропейски произход, така наречените евреи ашкенази. В тази група честотата на такива редки лизозомни заболявания като болестта на Гоше,
Tay-Sachs (амавротичен идиотизъм), Niemann-Pick, муколипидоза, с
висока честота на торсионна дистония, синдром на Блум
(една от генетичните форми на нанизъм, съчетана с повишена чувствителност към слънчева радиация, телеангиектазия, нарушена пигментация на кожата и предразположение към злокачествени новообразувания). Освен това, увеличаването на честотата на тези заболявания, като правило, се дължи на широкото разпространение на специфични мутации в съответните гени. Друг пример е автозомно-доминантната форма на болестта на Паркинсон, причинена от мутации в богатия на левцин киназа 2 ген – LRRK2. При европейските пациенти с фамилни форми на заболяването се среща специфична мутация в гена LRRK2 (G2019S) с честота 6%, докато при същите пациенти, но евреи ашкенази, честотата на тази мутация достига 30-40%.
Определени полиморфни мутации в два гена, свързани с рак на гърдата и рак на яйчниците, са широко разпространени в тази етническа група. Дори такава добре позната болест като кистозна фиброза
Ашкеназките евреи до голяма степен се обясняват с наличието на много специфична мутация (W128X). Имайте предвид, че в Израел цялостното изследване на бременни жени включва анализ на хетерозиготно носителство на мутации в някои от гените, отговорни за изброените по-горе заболявания.
Съвсем различен спектър от наследствени заболявания, срещащи се с повишена честота, се наблюдава сред финландците, тоест в друга изолирана етническа група. Открити са финландски специфични мутации за най-малко 30 различни моногенни заболявания.
Честотата на вродената нефроза при финландците достига 1:8000. Близо до
1% от местното население на Финландия са хетерозиготни носители на мутацията, която се среща в хомозиготно състояние в повече от
90% от пациентите с диастрофична дисплазия - една от формите на скелетна дисплазия, характеризираща се с тежка сколиоза, двустранна вродена деформация на ръцете и краката, удебеляване на ушите,
преждевременна калцификация на ребрените хрущяли, наличие, в
в повечето случаи цепнатини на твърдото небце. Две форми на наследствена офталмопатия се срещат с повишена честота сред финландците,
всяка от които е причинена от специфична мутация. Това е сгъната атрофия на хориоидеята и ретината, както и плоска роговица от тип II, при която се наблюдава помътняване на роговицата и паренхима на роговицата още в ранна детска възраст;
в този случай нивото на хиперметропия достига или дори надвишава +10D. СЪС
Инфантилната цероидна липофусциноза, фамилната амилоидоза, една от генетичните форми, се срещат с повишена честота във финландските популации
прогресивна миоклонична епилепсия (Unverricht-Lundborg).
Примери за такива генетични изолати не са изолирани.
Като цяло, разпространението на различни мутации в популациите зависи от две сили, действащи в различни посоки - честотата на възникване на мутациите и отрицателната или положителна селекция по отношение на техните носители. Например, мутация, която има отрицателен ефект върху жизнеспособността в хомозиготно състояние, може да стане широко разпространена в популацията, ако осигурява някои предимства в хетерозиготно състояние. Класическият пример е мутация в гена на β-глобина, която, когато е хомозиготна, води до сърповидноклетъчна анемия. Мутантният хемоглобин има намалена разтворимост и повишена способност за полимеризация, в
В резултат червените кръвни клетки на пациентите придобиват сърповидна форма. Такива червени кръвни клетки губят своята пластичност, запушват малките съдове и се хемолизират. След това се развиват огнища на исхемия и инфаркт във вътрешните органи, гръбначния и главния мозък. Заболяването често се среща в Централна Африка, Индия, средиземноморските страни,
Близък и Среден изток, включително Азербайджан, Узбекистан и Армения. Оказа се, че в същите региони на света е широко разпространен маларийният плазмодий, който причинява сериозно инфекциозно заболяване - малария.
Хетерозигните носители на мутации в β-глобиновия ген имат повишена резистентност към малария. Честотата на хетерозиготно носителство на мутации в β-глобиновия ген в тези популации достига 5-8%.
Комбинацията от горните фактори води до популационен полиморфизъм, тоест стабилното съвместно съществуване на няколко генетични форми в рамките на една и съща популация, докато различните популации могат да се различават по нива или честоти на полиморфизъм. Важна характеристика на индивид с определен генотип е неговата
фитнес (W), тоест вероятността да оцелееш до репродуктивна възраст и да оставиш потомство. Обща годност на населението
е средната стойност на годността на всички индивиди и нейното нормализирано отклонение от максимално възможната стойност – (Wmax –
W)/Wmax – определя генетично натоварваненаселение, което е средна мярка за разпространението в популация на мутации, които намаляват годността на индивидите. Той определя дела в общата популация на хомозиготни и хетерозиготни носители на мутации, които имат отрицателен ефект върху жизнеспособността. Естествените популации на растения и животни, както и хората, са обременени с различни мутации.
По отношение на хората, генетичният товар определя разпространението в различни популации на мутации, свързани с наследствена патология. Постоянно се появяват доминиращи мутации
Някои рецесивни мутации се откриват в редки хомозиготи, но основният дял от генетичния товар, като айсберг, е скрит в генофонда на популацията в хетерозиготно състояние. Според изключителния руски генетик S.S.
Четвериковските мутации в естествените популации представляват еволюционния резерв на вида. Това направление на популационната генетика се развива интензивно в нашата страна през първата половина и средата на миналия век в трудовете на Г. Мелер, Н. П. Дубинин, а след това Р. Л. Берг, М. Д. Голубовски и др в популации, включително тези, които водят до летален ефект в хомозиготно състояние,
достига няколко десетки процента и съставът на тези мутации непрекъснато се променя и различни специфични мутации стават широко разпространени в различни години.
IN В заключение подчертаваме товапопулационно генетично
изследването е от първостепенно значение за провеждане
епидемиологични изследвания с цел правилна организация на медицинските
генетично консултиране на населението и профилактика на наследствена патология.
Глава 1.7. Структура на субстанцията на наследствеността - ДНК
Неврологичните и психичните разстройства представляват 13% от глобалното бреме на болестите, пряко засягащи повече от 450 милиона души по света. Разпространението на тези заболявания вероятно ще продължи да нараства в резултат на нарастващата продължителност на живота на населението. За съжаление почти половината от пациентите с шизофрения понастоящем не получават подходящи медицински грижи, отчасти защото ранните симптоми на шизофрения често се бъркат с тези, наблюдавани при други психични разстройства (напр. психотична депресия или биполярно разстройство). Други заболявания, като синдром на Rett (RTT) и неврофиброматоза тип II (NF2), изискват мултидисциплинарен подход и лечение в специализирани медицински центрове. В допълнение, повечето от тези разстройства са комплексни, резултат от взаимодействието на генетични фактори и фактори на околната среда.
Въз основа на данни от двойни проучвания, наследствеността на някои психични разстройства е висока. Това се отнася за аутизма и шизофренията, с нива на наследственост съответно около 90% и 80%. Въпреки това, тези заболявания също често се срещат като изолирани случаи, само с едно засегнато дете, родено от незасегнати родители без фамилна анамнеза за заболяването. Едно от възможните обяснения за това явление е появата на мутации de novo, където мутации възникват по време на сперматогенеза или оогенеза (мутации на зародишна линия) и следователно присъстват при пациента, но не се откриват при незасегнатия родител. Този генетичен механизъм наскоро беше в центъра на вниманието при обясняване на част от генетичната основа на нарушенията в развитието на мозъчната система.
Като се има предвид фактът, че се смята, че човешкият геном съдържа приблизително 22 333 гена, може да се каже, че повече от 17 800 гена се експресират в човешкия мозък. Мутациите, засягащи почти всеки от тези гени, в комбинация с факторите на околната среда, могат да допринесат за развитието на неврологични и психиатрични разстройства на мозъка. Последните проучвания идентифицираха редица причинно-следствени генни мутации и показаха значителната роля, която генетиката играе при неврологични и психиатрични разстройства. Тези проучвания показват участието на редки (<1% частоты) точечных мутаций и вариаций числа копий (CNVs, то есть геномных делеций или дублирования от>1 kb до няколко Mb), които могат да се появят в региони без гени или които могат да засегнат единичен ген или да включват непрекъснат набор от гени в генетичната етиология на аутизъм, шизофрения, интелектуално увреждане, разстройство с дефицит на вниманието и други невропсихиатрични разстройства .
Отдавна е известно, че неврологичните и психиатрични разстройства се срещат в семейства, което предполага наследственост с основен генетичен компонент на заболяването. За някои неврологични заболявания, като NF2 или RTT, е установена генетична причина. Въпреки това, за по-голямата част от неврологичните и психиатрични разстройства, като шизофрения, аутизъм, биполярно разстройство и синдром на неспокойните крака, генетичните причини остават до голяма степен неизвестни. Последните разработки в технологиите за секвениране на ДНК откриха нови възможности за нашето разбиране на генетичните механизми, които са в основата на тези разстройства. С помощта на масивни паралелни платформи за секвениране на ДНК (наричани още „следващо поколение“) могат да се търсят мутации във всички гени на генома на човек в една проба (експеримент).
Известна стойност Де Новомутации (т.е. придобити мутации в потомството) при психични разстройства като умствена изостаналост (ID), аутизъм и шизофрения. Наистина, в много скорошни геномни проучвания анализът на геномите на засегнатите индивиди и сравнението им с геномите на родителите показа, че редки кодиращи и некодиращи вариации de novoзначително свързани с риска от аутизъм и шизофрения. Предполага се, че големият брой нови случаи на тези заболявания са причинени отчасти от мутации ново,което може да компенсира алелните загуби поради силно намален репродуктивен капацитет, като по този начин поддържа висока честота на тези заболявания. Изненадващо е, че мутациите de novoса доста чести (от порядъка на 100 нови мутации на дете), като само няколко (от порядъка на една на дете) в кодиращи региони.
Мутации de novoвъншни кодиращи региони, като например в промоторни, интронични или междугенни региони, също могат да бъдат свързани с болест. Предизвикателството обаче е да се определи коя от тези мутации е патогенна.
Трябва да се вземат предвид няколко основни линии на доказателства, когато се оценява патогенността на наблюдаваното Де Новомутации: Де Новостепен на мутация, генна функция, влияние на мутацията и клинични корелати. Ключовите въпроси сега могат да бъдат формулирани по следния начин: Колко гена ще участват в неврологични и психиатрични разстройства? Какви специфични генни пътища са включени? Какви са последствията от мутациите de novoза генетична консултация? На тези въпроси трябва да се отговори, за да се подобри диагнозата и да се разработят лечения.
Ролята на мутациите de novoпри човешки заболявания е добре известно, особено в областта на раковата генетика и доминиращите менделски разстройства като синдромите на Кабуки и Шинцел-Гиедион. И двата синдрома се характеризират с тежка интелектуална недостатъчност и вродени лицеви аномалии и наскоро беше доказано, че са причинени от мутации de novo V MLL2 гениИ SETBP1, съответно. Наскоро изследванията на Сандърс и др., Нийл и др., О'Роук и др. потвърди приноса Де Новомутации в етиологията на аутизма. Всяко изследване идентифицира списък с мутации ново,присъства в пробандите, но само няколко гена са идентифицирани с няколко de novo (CHD8, SCN2A, KATNAL2И NTNG1). Взаимодействието на протеини и базираните на пътя анализи от тези проучвания показват значителна свързаност и общ биологичен път между гени, носещи мутации de novoв случаи на аутизъм. Протеиновите мрежи, участващи в ремоделирането на хроматина, убиквитинирането и развитието на невроните, са идентифицирани като потенциални мишени на гени за чувствителност към аутизъм. И накрая, тези проучвания показват, че 1000 или повече гена могат да се тълкуват като имащи всеобхватни мутации, които допринасят за аутизма.
Технологичният напредък в секвенирането на ДНК по същество революционизира изследването на генетичните вариации в човешкия геном и даде възможност за идентифициране на много видове мутации, включително замествания на единични базови двойки, инсерции/делеции, CNV, инверсии и повторни експанзии, както и тези, които се считат за соматични и зародишни мутации. Доказано е, че всички тези видове мутации играят роля в човешките заболявания. Изглежда, че единичните нуклеотидни мутации са предимно от „бащин произход“, докато делециите могат да бъдат предимно от „майчин произход“. Това може да се обясни с разликите между мъжката и женската гаметогенеза. Например, в изследване на неврофиброматоза, 16 от 21 мутации се състоят от делеции от майчин произход, а 9 от 11 точкови мутации са от бащин произход.
Различни видове мутации могат да се предават от родител на дете или да се придобиват спонтанно. Механизмът, задвижващ последното, привлече вниманието през последните години поради значението на този тип мутация при заболявания като шизофрения и аутизъм. Скорост на мутация ново,изглежда доминира с възрастта на бащата. Процентът тук се увеличава с възрастта на бащата, вероятно поради последиците от намалената ефективност на репликацията на ДНК или механизмите за възстановяване, които се очаква да намаляват с възрастта. Следователно рискът от заболяването трябва да нараства с увеличаване на възрастта на бащата. Установено е, че това се случва в много случаи, включително синдром на Crouzon, множествена ендокринна неоплазия тип II и неврофиброматоза тип I. Съвсем наскоро O'Roak и др. наблюдава забележим бащин компонент в 51 мутации ново,идентифицирани чрез изследване на последователността на 188 родители-деца със случаи на спорадичен аутизъм. Тези резултати са подобни на наблюдаваните в последните доклади за CNNн новос интелектуално увреждане. Тази корелация може да се обясни със значително по-големия брой митотични клетъчни деления в зародишните клетки или сперматоцитите преди мейозата през целия живот на мъжете в сравнение с това, което се случва по време на оогенезата при жените.
Въз основа на установения брой клетъчни деления, които се случват в оогенезата (от раждането до менопаузата) в сравнение със сперматогенезата (от пубертета до края на живота), Джеймс Ф. Кроу изчислява, че на 30-годишна възраст средният брой хромозомни повторения от зиготата до спермата производството е 16,5 пъти по-високо, отколкото от зиготата до образуването на яйцеклетката.
Генетичният мозаицизъм се причинява от появата de novoмитотични мутации, се проявява много рано в ембрионалното развитие и се определя като наличието на множество клетъчни клонинги със специфичен генотип в един и същ индивид. Съществува соматичен и зародишен мозаицизъм, но зародишният мозаицизъм може да допринесе за предаването на това, което може да се предаде чрез мутация de novoпотомство.
Спонтанните мутации, възникващи в соматичните клетки (по време на митоза, след оплождане), също могат да играят роля в генезиса на заболявания, свързани с нарушения в развитието.