این سندرم به دلیل عدم وجود بخشی از ماده ژنتیکی واقع در بازوی کوتاه کروموزوم 11 رخ می دهد. حذف بخشی از ماده ژنتیکی حذف نامیده می شود. حذف منجر به شکست آن دسته از عملکردهایی می شود که قرار بود توسط ژن های از دست رفته انجام شوند.
همه ژن ها، به استثنای برخی که روی کروموزوم های جنسی قرار دارند، در دو نسخه ارائه می شوند. هر فرد یک بخش از ژن ها را از مادر خود دریافت می کند و بخش دوم مشابه را از پدر خود دریافت می کند. آنها به نوبه خود جفت ژن های خود را از والدین خود دریافت کردند. مواد ژنتیکی از طریق سلول های تولید مثلی از والدین منتقل می شود. سلول های جنسی (تخمک یا اسپرم) تنها سلول هایی در بدن هستند که تنها یک کپی از مواد ژنتیکی را حمل می کنند. قبل از اینکه ماده ژنتیکی وارد شود سلول جنسیبین دو نسخه از ژن ها، ژن ها به هم ریخته می شوند و هر یک از والدین مواد ژنتیکی را در سلول تولیدمثلی قرار می دهند، که ترکیبی از موادی است که به نوبه خود از والدین خود دریافت کرده است. زندگی جدیدآنها نیز قبل از قرار گرفتن در سلول تولیدمثلی برای ایجاد نسل بعدی، مخلوط می شوند. به این فرآیند عبور دادن می گویند. بین نواحی همولوگ کروموزوم ها در طول تشکیل سلول های زایا رخ می دهد. از طریق فرآیند عبور، ژن ها می توانند ترکیبات جدیدی ایجاد کنند. این اختلاط تنوع نسل های جدید را تضمین می کند. این برای چیست؟ این امر به منظور اطمینان از تنوع نسلی، ضروری است در غیر این صورتما نسخه های دقیقی از کروموزوم های دریافتی از یکی از والدین خود را به فرزندان خود می دهیم، تنوع نسلی بسیار محدود خواهد بود، که باعث می شود تکامل بیولوژیکیروی زمین بسیار دشوار خواهد بود و بنابراین شانس بقا را کاهش می دهد. در لحظه ای که چنین فرآیندهایی اتفاق می افتد، یک قطعه از کروموزوم می تواند جدا شود و یک "حذف" ایجاد شود. حذف نوعی جهش است. اگر برای اولین بار رخ دهد، چنین جهشی را جهش de novo (اولین، اولیه) می نامند. علاوه بر جهش هایی که برای اولین بار در بدن ظاهر شد، جهش هایی وجود دارد که ارثی هستند. یک جهش de novo می تواند به نسل های بعدی منتقل شود، در این مرحله دیگر جهش de novo نامیده نمی شود.
در سندرم WAGR بخشی از کد ژنتیکی حذف شده و مواد ژنتیکی کافی وجود ندارد.
در طبیعت، شرایط مخالف وجود دارد، زمانی که بیماری خود را به دلیل کپی اضافی از مواد ژنتیکی نشان می دهد.
تظاهرات سندرم WAGR بستگی به این دارد که کدام ژن در نتیجه حذف خاموش شود. ژن های همسایه همیشه از بین می روند. در WAGR، ژن PAX6 و ژن WT1 همیشه از بین می روند که منجر به تظاهرات معمولی بیماری می شود. جهش های نقطه ای در ژن PAX6 منجر به آنیریدیا و جهش در WT1 منجر به تومور ویلمز می شود. با WAGR، هیچ جهشی در این ژن ها وجود ندارد - خود ژن ها وجود ندارند.
افراد مبتلا به سندرم WAGRO (حرف O اضافه شده است - چاقی) به ژن BDNF آسیب می زند. این ژن در مغز بیان می شود و در زندگی نورون ها مهم است. پروتئین تولید شده تحت تأثیر این ژن به احتمال زیاد در تنظیم سیری، تشنگی و وزن بدن نقش دارد. از دست دادن BDNF به احتمال زیاد با چاقی مرتبط است که در ابتدا شروع می شود دوران کودکیدر کودکان مبتلا به سندرم WAGRO بیماران مبتلا به WAGRO در معرض خطر بیشتری برای مشکلات عصبی مانند کاهش هوش و اوتیسم هستند. به طور کامل بررسی نشده است که آیا این خطر به طور خاص با از دست دادن ژن BDNF مرتبط است یا خیر
ما چیزی در مورد ژن هایی که در سندرم WAGR خاموش می شوند می دانیم:
WT1
WT1 یک ژن (ژن تومور ویلمز) است که پروتئین لازم را ترشح می کند توسعه طبیعیکلیه ها و غدد جنسی (تخمدان در زنان و بیضه در مردان). در این بافت ها پروتئین در تمایز سلولی و آپوپتوز نقش دارد. برای انجام همه اینها، WT1 برای تنظیم فعالیت سایر ژن ها با اتصال به مناطق DNA عمل می کند.
ژن WT1 برای سرکوب تومور ویلمز مورد نیاز است. گونه ای از نام ژن Wilm's tumor suppressor gene1 (ژن سرکوب کننده توسعه تومور ویلمز) وجود دارد. جهش یا عدم وجود آن منجر به افزایش خطر توسعه تومور می شود. دقیقاً به دلیل احتمال درگیری این ژن است. در سندرم WAGR که نظارت دائمی بر وضعیت کلیه ها ضروری است.
PAX6
PAX6 متعلق به خانوادهای از ژنها است که نقش مهمی در رشد اندامها و بافتها دارند. رشد جنینی. اعضای خانواده PAX برای عملکرد طبیعی سلول های مختلف بدن پس از تولد مهم هستند. ژنهای خانواده PAX در سنتز پروتئینهایی نقش دارند که بخشهای خاصی از DNA را به هم متصل میکنند و در نتیجه فعالیت ژنهای دیگر را کنترل میکنند. به دلیل این خاصیت، پروتئین های PAX را فاکتورهای رونویسی می نامند.
در طول رشد جنینی، پروتئین PAX 6 ژن های دخیل در رشد چشم، مغز، نخاع و پانکراس را فعال می کند. PAX 6 در توسعه شرکت دارد سلول های عصبیدستگاه بویایی که مسئول حس بویایی است. در حال حاضر ویژگی PAX 6 در طول رشد داخل رحمیبه احتمال زیاد به طور کامل مطالعه نشده است و به مرور زمان حقایق جدیدی دریافت می کنیم. پروتئین PAX6 پس از تولد، بسیاری از ژن ها را در چشم تنظیم می کند.
عدم عملکرد ژن PAX 6 منجر به مشکلات چشمی پس از تولد می شود.
BDNF
ژن BDNF پروتئینی را کد می کند که در مغز و نخاع. این ژن در رشد و بلوغ سلول های عصبی نقش دارد. پروتئین BDNF در سیناپس های مغز فعال است. سیناپس ها می توانند در پاسخ به تجربه تغییر کرده و سازگار شوند. پروتئین BDNF به تنظیم تنوع سیناپسی کمک می کند که برای یادگیری و حافظه بسیار مهم است.
پروتئین BDNF در مناطقی از مغز یافت می شود که سیری، تشنگی و وزن بدن را کنترل می کند. به احتمال زیاد، این پروتئین به این فرآیندها کمک می کند.
بیان این ژن در بیماری های آلزایمر، پارکینسون و هانتینگتون کاهش می یابد و این ژن ممکن است در پاسخ به استرس و بیماری های اختلال خلقی نقش داشته باشد. ژن BDNF توجه بسیاری از محققان را به خود جلب کرده است. مطالعاتی وجود دارد که فعالیت پروتئین BDNF در مغز را بسته به این موضوع مطالعه می کند تمرین فیزیکی، رژیم های غذایی، استرس روانیو شرایط دیگر فعالیت این پروتئین با فعالیت ذهنی و شرایط روحی، تلاش می شود تا بر سطح آن تأثیر بگذارد.
ممنون میشم اگه راهنماییم کنید اطلاعات جدیددر مورد این سوال همه چیز را در نظرات بنویسید.
توجه داشته باشید:
کلمات پروتئین و پروتئین مترادف هستند
E.V. توزلیان، فوق تخصص غدد کودکان، متخصص ژنتیک، دکتری تخصصی، جدا زیرمجموعه ساختاری"موسسه تحقیقاتی بالینی اطفال" موسسه آموزشی بودجه دولتی آموزش عالی حرفه ای دانشگاه ملی تحقیقات پزشکی روسیه به نام. N.I. وزارت بهداشت پیروگوف فدراسیون روسیه، مسکو کلید واژه ها
: کودکان، سندرم نونان، تشخیص.
کلید واژه ها: کودکان، سندرم نونان، تشخیص.
مقاله سندرم نونان (سندرم اولریش-نونان، سندرم ترنروئید با کاریوتایپ طبیعی) را توصیف می کند - یک بیماری نادر آسیب شناسی مادرزادی، به صورت اتوزومال غالب به ارث می رسد، خانوادگی است، اما موارد پراکنده نیز رخ می دهد. این سندرم وجود یک ویژگی فنوتیپ سندرم شرشفسکی-ترنر را در افراد زن و مرد با کاریوتایپ طبیعی فرض میکند. ارائه شده توسط مشاهده بالینی. مشکلات جستجوی تشخیصی افتراقی و عدم آگاهی پزشکان در مورد این سندرمو اهمیت رویکرد میان رشته ای.
حقایق تاریخی
سندرم غیر معمول اولین بار توسط O. Kobylinski در سال 1883 ذکر شد (عکس 1).
قدیمی ترین شناخته شده مورد بالینیسندرم نونان که در سال 1883 توسط O. Kobylinski شرح داده شد
این بیماری در سال 1963 توسط متخصص قلب آمریکایی ژاکلین نونان توصیف شد که 9 بیمار مبتلا به تنگی دریچه را گزارش کرد. شریان ریوی، کوتاهی قد، هایپرتلوریسم، کاهش متوسط هوش، پتوز، کریپتورکیدیسم و ناهنجاری های اسکلتی. دکتر نونان، که به عنوان متخصص قلب کودکاندر دانشگاه آیووا، متوجه شد که کودکان با نوع کمیاببیماری قلبی - تنگی دریچه ریوی - معمولی ناهنجاری های فیزیکیبه شکل قد کوتاه، گردن بال مانند، چشم های پهن و گوش های کم قرار دارد. پسران و دختران به یک اندازه تحت تأثیر قرار گرفتند. دکتر جان اوپیتز، دانش آموز سابقنونان اولین کسی بود که اصطلاح "سندرم نونان" را برای توصیف وضعیت کودکانی که علائمی شبیه به آنچه که توسط نونان توصیف شده بود را ابداع کرد. نونان بعداً مقاله ای با عنوان «هیپرتلوریسم با فنوتیپ ترنر» نوشت و در سمپوزیومی در سال 1971 بیماری های قلبی عروقینام "سندرم نونان" رسما به رسمیت شناخته شد.
اتیولوژی و پاتوژنز
سندرم نونان یک اختلال اتوزومال غالب با بیان متغیر است (شکل 1). ژن سندرم نونان محلی است شانه بلندکروموزوم 12. ناهمگونی ژنتیکی این سندرم را نمی توان رد کرد. پراکنده و اشکال خانوادهسندرم با فرم ارثی اتوزومال غالب. در موارد خانوادگی، ژن جهش یافته، به طور معمول، از مادر به ارث می رسد، زیرا به دلیل رذایل شدیدتوسعه سیستم تناسلی ادراریمردان مبتلا به این بیماری اغلب نابارور هستند. اکثر موارد گزارش شده پراکنده هستند که در اثر جهش های نو ایجاد می شوند.

. نوع توارث اتوزومال غالب
ترکیبات توصیف شده از سندرم نونان با نوروفیبروماتوز نوع I در چندین خانواده منجر به این فرض شد اتصال احتمالیدو جایگاه مستقل 17q11.2 کروموزوم 17. برخی از بیماران دارای ریزحذفات در جایگاه 22q11 کروموزوم 22 هستند. در این موارد تظاهرات بالینیسندرم نونان با کم کاری تیموس و سندرم دی جورج ترکیب می شود. تعدادی از نویسندگان در مورد مشارکت ژن های احتمالی لنفوژنز در پاتوژنز سندرم به دلیل وجود ناهنجاری های صورت و جسمی مشابه سندرم ترنر و فراوانی بالای آسیب شناسی بحث می کنند. سیستم لنفاوی.
اکثر دلیل مشترکسندرم نونان یک جهش در ژن PTPN11 است که تقریباً در 50 درصد بیماران یافت می شود. پروتئین کدگذاری شده توسط ژن PTPN11 متعلق به خانواده ای از مولکول ها است که پاسخ را تنظیم می کنند. سلول های یوکاریوتیبه سیگنال های خارجی بزرگترین عددجهشهای سندرم نونان در اگزونهای 3، 7 و 13 ژن PTPN11 قرار دارند و دامنههای پروتئینی مسئول انتقال پروتئین به حالت فعال.
ایده های احتمالی در مورد پاتوژنز با مکانیسم های زیر نشان داده می شود:
مسیر RAS-MAPK بسیار است مسیر مهمانتقال سیگنال، که از طریق آن لیگاندهای خارج سلولی - فاکتورهای رشد خاص، سیتوکین ها و هورمون ها - تحریک می شوند. تکثیر سلولیتمایز، بقا و متابولیسم (شکل 2). پس از اتصال لیگاند، گیرنده های سطح سلولی در مکان هایی در ناحیه آندوپلاسمی خود فسفریله می شوند. این اتصال شامل پروتئینهای آداپتور (به عنوان مثال، GRB2) است که یک کمپلکس سازنده را با فاکتورهای تبادل نوکلئوتیدی گوانین (مانند SOS) تشکیل میدهند که RAS غیرفعال متصل به GDP را به شکل فعال متصل به GTP تبدیل میکند. سپس پروتئین های RAS فعال شده، آبشار RAF-MEKERK را از طریق یک سری واکنش های فسفوریلاسیون فعال می کنند. در نتیجه، ERK فعال شده وارد هسته میشود تا رونویسی ژنهای هدف را تغییر دهد و فعالیت اهداف آندوپلاسمی را برای القای پاسخهای سلولی کوتاهمدت و بلندمدت کافی به محرک تنظیم میکند. تمام ژن های دخیل در سندرم نونان پروتئین های جدایی ناپذیر این مسیر و جهش ها را رمزگذاری می کنند. ایجاد بیماری، معمولا سیگنال عبوری از این مسیر را تقویت می کند.
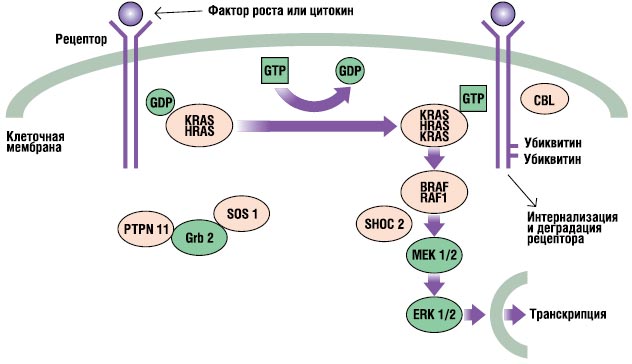
. مسیر سیگنالینگ RAS-MAPK. سیگنال های رشد از گیرنده های فعال شده با فاکتور رشد به هسته منتقل می شوند. جهش در PTPN11، KRAS، SOS1، NRAS و RAF1 با سندرم نونان همراه است و جهش در SHOC2 و CBL با یک فنوتیپ شبیه به سندرم نونان مرتبط است.
ویژگی های بالینی سندرم نونان
فنوتیپ بیماران مبتلا به سندرم نونان شبیه به سندرم ترنر است: گردن کوتاه با چین های ناخنک یا رشد کم مو، قد کوتاه، هیپرتلوریسم شقاق های کف دست (شکل 2). میکروآنومالی های صورت شامل شقاق کف دستی آنتی منگولوئید، کانتوس بیرونی رو به پایین، پتوز، اپیکانتوس، گوش های کم تنظیم، مارپیچ تا شده است. گوش هامال اکلوژن، شکاف یوولا کام نرمکام گوتیک، میکروگناتیا و میکروژنیا. قفسه سینه تیروئیدی شکل با نوک سینه های هیپوپلاستیک و با فاصله زیاد است، جناغ در قسمت فوقانی بیرون زده و در قسمت پایین فرو می رود. حدود 20 درصد از بیماران آسیب شناسی اسکلتی متوسط دارند. رایج ترین تغییر شکل قیف قفسه سینهکیفوز، اسکولیوز؛ کمتر - کاهش تعداد مهره های گردنی و همجوشی آنها، یادآور ناهنجاری های سندرم کلیپل-فیل.

. فنوتیپ های سندرم نونان
بیماران مبتلا به سندرم نونان معمولا موهای بلوند، ضخیم و مجعد با رشد غیرعادی در تاج سر دارند. نقاط تاریکروی پوست، هیپرتریکوزیس، دیستروفی صفحات ناخن، ناهنجاری در رویش و محل دندان ها، تمایل به ایجاد اسکارهای کلوئیدی، افزایش انبساط پوست. یک سوم بیماران مبتلا به لنف ادم محیطی هستند؛ بیشتر اوقات، لنف ادم دست و پا در کودکان رخ می دهد. سن پایین. یک علامت رایج آسیب شناسی بینایی (نزدیک بینی، استرابیسم، اگزوفتالموس متوسط و غیره) است. تأخیر رشد تقریباً در 75 درصد بیماران رخ می دهد، در پسران بارزتر است و معمولاً ناچیز است. تأخیر رشد در سالهای اول زندگی خود را نشان می دهد؛ کمتر اوقات، کمبودهای جزئی در قد و وزن هنگام تولد مشاهده می شود. از ماه های اول زندگی کاهش اشتها مشاهده می شود. سن استخوانیمعمولا از پاسپورت عقب می ماند.
یکی از ویژگی های این سندرم کریپتورکیدیسم یک طرفه یا دو طرفه است که در 70-75٪ از بیماران مرد رخ می دهد؛ در بیماران بزرگسال، آزواسپرمی، اولیگواسپرمی، تغییرات دژنراتیوبیضه ها با این وجود، بلوغ خود به خود و گاهی با تاخیر اتفاق می افتد. دختران اغلب با تاخیر در شکل گیری قاعدگی مواجه می شوند و گاهی اوقات بی نظمی نیز وجود دارد چرخه قاعدگی. باروری ممکن است در بیماران هر دو جنس طبیعی باشد.
عقب ماندگی ذهنی در بیش از نیمی از بیماران، معمولا جزئی تشخیص داده می شود. ویژگی های رفتاری، عدم بازداری و اختلال کمبود توجه اغلب ذکر شده است. گفتار معمولاً بهتر از سایر حوزه های فکری توسعه می یابد. میزان کاهش هوش با شدت اختلالات جسمی ارتباطی ندارد [Marincheva G.S.، 1988]. در موارد جداگانه، ناهنجاری های مرکزی سیستم عصبی(هیدروسفالوس، اسپینا بیفیدا)، انفارکتوس مغزی ترومبوآمبولیک، احتمالاً با هیپوپلازی عروقی همراه است.
رذایل اعضای داخلیبا سندرم نونان کاملا مشخص است. معمول ترین ناهنجاری های قلبی عروقی عبارتند از: تنگی دریچهشریان ریوی (حدود 60٪ از بیماران)، کاردیومیوپاتی هیپرتروفیک(20-30%)، ناهنجاری های ساختاری دریچه میترالنقايص ديواره بين دهليزي، تترالوژي فالوت; کوآرکتاسیون آئورت فقط در بیماران مرد توصیف شده است.
در یک سوم بیماران، نقایص سیستم ادراری ثبت می شود (هیپوپلازی کلیه، تکثیر لگن، هیدرونفروز، مگاورتر و غیره).
اغلب، با سندرم نونان، افزایش خونریزی مشاهده می شود، به ویژه زمانی که مداخلات جراحی V حفره دهانو نازوفارنکس شناسایی می شوند عیوب مختلفانعقاد: نارسایی سیستم پلاکتی، کاهش سطح فاکتورهای انعقادی به ویژه XI و XII، افزایش زمان ترومبوپلاستین. گزارش هایی از ترکیب سندرم نونان با لوسمی و رابدومیوسارکوم وجود دارد که ممکن است نشان دهنده افزایش خفیف خطر بدخیمی در این بیماران باشد.
جدول 1 ویژگی های فنوتیپ در سندرم نونان را نشان می دهد که با افزایش سن بیمار تغییر می کند. جدول 2 همبستگی بین فنوتیپ و ژنوتیپ را در سندرم نونان نشان می دهد.
میز 1. ویژگی های معمولی صورت بیماران مبتلا به سندرم نونان بر اساس سن
| پیشانی، صورت، مو | چشم ها | گوش ها | بینی | دهان | گردن | |
| نوزاد* | پیشانی بلند، خط موی کم در پشت سر | هایپرتلوریسم، شیب رو به پایین شکاف های کف دست، چین اپیکانتوس | – | ریشه فرورفته کوتاه و پهن، نوک رو به بالا | فیتروم عمیق فرورفته، قله های پهن و بلند لبه قرمز لب، میکروگناتیا | پوست اضافی در پشت سر |
| نوزاد (2 تا 12 ماه) | سر بزرگ، پیشانی بلند و بیرون زده | هایپرتلوریسم، پتوز یا افتادگی پلک ضخیم | – | ریشه فرورفته کوتاه و پهن | – | – |
| کودک (1-12 سال) | ویژگی های خشن، صورت دراز | – | – | – | – | – |
| نوجوان (12 تا 18 سال) | صورت میوپاتیک | – | – | پل بلند و نازک است | – | تشکیل آشکار چین های دهانه رحم |
| بزرگسالان (بیش از 18 سال) | ویژگی های متمایز صورت اصلاح می شود، پوست نازک و شفاف به نظر می رسد | – | – | چین بینی بیرون زده | – | – |
| تمام سنین | – | عنبیه آبی و سبز، ابروهای الماسی شکل | گوش های پایین و چرخشی به عقب با چین های ضخیم | – | – | – |
جدول 2. ارتباط بین ژنوتیپ و فنوتیپ در سندرم نونان*
| سیستم قلبی عروقی | ارتفاع | توسعه | پوست و مو | دیگر | |
| PTPN11 (تقریباً 50%) | تنگی تنه ریوی بارزتر است. کمتر - کاردیومیوپاتی هیپرتروفیک و نقص سپتوم دهلیزی | ارتفاع کوتاه تر؛ غلظت IGF1 پایین تر | بیماران مبتلا به N308D و N308S دارند کاهش جزئییا هوش عادی | – | بارزتر دیاتز هموراژیکو لوسمی میلومونوسیتی نوجوانان |
| SOS1 (تقریباً 10%) | نقص سپتوم دهلیزی کمتر | رشد بلندتر | کاهش کمتر در هوش، تاخیر در رشد گفتار | شبیه به سندرم قلبی - صورت | – |
| RAF1 (تقریباً 10%) | کاردیومیوپاتی هیپرتروفیک شدیدتر | – | – | بیشتر خال های مادرزادی, لنتیگو, کافه au lait spots | – |
| کراس (<2%) | – | – | تاخیر شناختی شدیدتر | شبیه به سندرم قلبی - صورت | – |
| NRAS (<1%) | – | – | – | – | – |
داده های حاصل از مطالعات آزمایشگاهی و عملکردی
هیچ نشانگر بیوشیمیایی خاصی برای تشخیص سندرم نونان وجود ندارد. در برخی از بیماران، کاهش ترشح خودبهخودی هورمون رشد در شب با پاسخ طبیعی به آزمایشهای محرک دارویی (کلونیدین و آرژنین)، کاهش سطح سوماتومدین-C و کاهش پاسخ سوماتومدینها به تجویز دارو مشاهده میشود. هورمون رشد.
معیارهای تشخیص
تشخیص سندرم نونان بر اساس علائم بالینی انجام می شود، در برخی موارد تشخیص با نتایج یک مطالعه ژنتیک مولکولی تایید می شود. معیارهای تشخیص سندرم شامل وجود چهره مشخص (با کاریوتایپ طبیعی) همراه با یکی از علائم زیر است: آسیب شناسی قلبی، کوتاهی قد یا کریپتورکیدیسم (در پسران)، بلوغ تاخیری (در دختران). برای شناسایی آسیب شناسی قلبی عروقی، انجام یک معاینه اولتراسوند قلب با تعیین پویا اندازه حفره ها و دیواره بطن ها ضروری است. تشخیص پیش از تولد این بیماری با استفاده از مانیتورینگ اولتراسوند امکان پذیر است که امکان شناسایی نقص های قلبی و ناهنجاری های ساختار گردن را فراهم می کند.
تشخیص های افتراقی
در دختران، تشخیص افتراقی در درجه اول سندرم ترنر است. یک مطالعه سیتوژنتیک می تواند تشخیص را روشن کند. علائم فنوتیپی سندرم نونان در تعدادی دیگر از بیماری ها دیده می شود: سندرم ویلیامز، سندرم LEOPARD، سندرم دوبوویتز، سندرم قلب و صورت، کورنلیا د لانگ، کوهن، روبینشتاین طیبی و غیره که شناسایی دقیق این بیماری ها تنها با انجام مول امکان پذیر خواهد بود. مطالعات ژنتیکی هر سندرم با مواد بالینی قابل توجهی که در حال حاضر به طور فعال در حال توسعه است.
رفتار
درمان بیماران مبتلا به سندرم نونان با هدف از بین بردن نقایص سیستم قلبی عروقی، عادی سازی عملکردهای ذهنی، تحریک رشد و رشد جنسی انجام می شود. برای درمان بیماران مبتلا به دیسپلازی دریچه ریوی، در میان روش های دیگر، بالون valvuloplasty با موفقیت استفاده می شود. برای تحریک رشد ذهنی از داروهای نوتروپیک و عروقی استفاده می شود. داروهایی با هدف تحریک رشد جنسی عمدتاً برای بیماران مبتلا به کریپتورکیدیسم تجویز می شوند. آماده سازی گنادوتروپین جفتی انسانی در دوزهای خاص سن استفاده می شود. در سنین بالاتر - در حضور هیپوگنادیسم - آماده سازی تستوسترون. در سال های اخیر، اشکال نوترکیب هورمون رشد انسانی در درمان بیماران مبتلا به سندرم نونان مورد استفاده قرار گرفته است. داده های بالینی با افزایش سطح سوماتومدین-C و پروتئین اتصال ویژه در طول درمان تأیید می شود. قد نهایی بیمارانی که به مدت طولانی تحت درمان با هورمون رشد قرار می گیرند، در برخی موارد از میانگین قد اعضای خانواده بیشتر است.
پیش بینی برای زندگی با شدت آسیب شناسی قلبی عروقی تعیین می شود.
جلوگیری بیماری بر اساس داده های مشاوره ژنتیک پزشکی است.
مشاوره ژنتیک پزشکی
هنگام انجام مشاوره ژنتیک پزشکی باید از نوع وراثت اتوزومال غالب و خطر بالای (50%) عود بیماری در خانواده دارای اشکال ارثی اقدام کرد. به منظور شناسایی ماهیت نوع وراثت، لازم است یک معاینه کامل از والدین انجام شود، زیرا این سندرم می تواند با حداقل علائم بالینی خود را نشان دهد. در حال حاضر، تشخیص ژنتیکی مولکولی بیماری توسعه یافته است و با تایپ کردن جهش در ژنهای PTPN11، SOS1، RAF1، KRAS، NRAS و غیره در حال بهبود است. روشهایی برای تشخیص قبل از تولد این بیماری در حال توسعه است.
مشاهده بالینی
Boy G.، 9 ساله (عکس 3)، توسط یک متخصص ژنتیک با تشخیص "آسیب شناسی کروموزومی؟، سندرم ویلیامز" (فنوتیپ خاص، ضخیم شدن برگچه های دریچه میترال، هیپرکلسمی هر 3 سال یک بار) در محل زندگی خود مشاهده شد. ?.

. ویژگی های فنوتیپ کودک مبتلا به سندرم نونان (اسکلت دراز صورت با گونه های چاق، گردن کوتاه، چین های ناخنک روی گردن، بینی کوتاه با سوراخ های بینی باز به سمت جلو، لب های چاق، چانه شیب دار، برش ضد مونگولوئید فیسسورپالپال مال اکلوژن، ماکروستومی)
شکایات برای کاهش حافظه، خستگی، کاهش نرخ رشد.
سابقه خانوادگی : والدین از نظر ملیت روسی هستند، از نظر خونی فامیل نیستند و خطرات شغلی ندارند، سالم هستند. قد پدر 192 سانتی متر و قد مادر 172 سانتی متر است و هیچ موردی از بیماری روانی، صرع و تاخیر رشدی در شجره نامه مشاهده نشد.
تاریخچه زندگی و بیماری : پسری از بارداری دوم (بارداری اول - m/a) که با تهدید به سقط جنین در تمام مدت همراه با پلی هیدرآمنیوس پیش رفت. تولد اول، به موقع، سریع، وزن هنگام تولد - 3400 گرم، طول - 50 سانتی متر. او بلافاصله فریاد زد، امتیاز آپگار - 7/9 امتیاز. در بدو تولد، متخصص نوزاد توجه را به فنوتیپ غیرمعمول کودک جلب کرد و یک مطالعه کاریوتیپ را توصیه کرد، نتیجه 46، XY (کاریوتیپ طبیعی مرد) بود. به کم کاری مادرزادی تیروئید مشکوک شد، پروفایل تیروئید بررسی شد و نتیجه وضعیت تیروئید طبیعی بود. سپس، کودک توسط یک متخصص ژنتیک با تشخیص احتمالی سندرم ویلیامز مشاهده شد. اوایل دوره پس از زایمان بدون ویژگی است. رشد حرکتی بر اساس سن، اولین کلمات - تا یک سال، گفتار عبارتی - در 2 سال و 3 ماه.
در سن 8 سالگی در مورد کاهش سرعت رشد، خستگی و کاهش حافظه توسط متخصص غدد مشاوره شد. معاینه اشعه ایکس از دست ها تاخیر متوسطی در سن استخوانی (BA) نسبت به سن پاسپورت (BA مربوط به 6 سال) بود. مطالعه پروفایل تیروئید افزایش متوسطی در هورمون محرک تیروئید با سطوح طبیعی T4 آزاد و سایر شاخص ها را نشان داد. سونوگرافی غده تیروئید - بدون آسیب شناسی. هورمون درمانی با مشاهده پویا بعدی تجویز شد.
با توجه به نامشخص بودن تشخیص در محل سکونت، متخصص ژنتیک کودک را به منظور روشن شدن تشخیص به مرکز مشاوره و تشخیصی منطقه ای مسکو برای کودکان فرستاد.
داده های تحقیق عینی:
قد - 126 سانتی متر، وزن - 21 کیلوگرم.
رشد فیزیکی پایین تر از حد متوسط، هماهنگ است. Sds رشد مطابق با -1 است (هنجار - 2+2). ویژگی های فنوتیپ (عکس 3): اسکلت صورت کشیده با گونه های چاق، گردن کوتاه، چین های ناخنک روی گردن، رشد کم مو در گردن، بینی کوتاه با سوراخ های بینی باز به جلو، لب های چاق، چانه شیب دار، برش ضد منگولوئید شقاق کف دست، مال اکلوژن، ماکروستومی، هیپرتلوریسم نوک سینه، عدم تقارن قفسه سینه، سینداکتیلی پوستی ناقص انگشتان دوم تا سوم روی پاها، بیش حرکتی شدید مفاصل بین فالانژیال، ناخن های شکننده و خشک. اندام های داخلی - بدون هیچ گونه ویژگی. رشد جنسی - Tanner I (که مربوط به دوره قبل از بلوغ است).
داده های تحقیقات آزمایشگاهی و عملکردی:
تجزیه و تحلیل بالینی خون و ادرار طبیعی است.
آزمایش خون بیوشیمیایی - شاخص ها در محدوده طبیعی هستند.
پروفایل تیروئید (TSH) - 7.5 µIU/ml (طبیعی - 0.4-4.0)، سایر شاخص ها طبیعی هستند.
هورمون سوماتوتروپیک (GH) - 7 نانوگرم در میلی لیتر (طبیعی - 7-10)، سوماتومدین-C - 250 نانوگرم در میلی لیتر (طبیعی - 88-360).
سونوگرافی غده تیروئید - بدون آسیب شناسی.
سونوگرافی اندام های داخلی - بدون هیچ ویژگی.
ECG - تاکی کاردی سینوسی، موقعیت طبیعی محور الکتریکی قلب.
EchoCG - درجه یک MVP با حداقل نارسایی، ضخیم شدن میکسوماتوز برگچه های دریچه میترال، وتر اضافی در حفره بطن چپ.
R-graphy از ستون فقرات – اسکولیوز سمت راست ستون فقرات قفسه سینه، درجه I.
R-graphy از دست ها با گرفتن ساعد - سن استخوان 7-8 سال.
هیچ الگوی EEG فعالیت صرعی ثبت نشد.
MRI مغز - بدون تغییر پاتولوژیک.
شنوایی - بدون آسیب شناسی.
تشخیص DNA: مطالعه ژنتیک مولکولی - هیچ حذفی از مکان های مورد مطالعه در ناحیه بحرانی کروموزوم 7 شناسایی نشد. جهش Gly434Ary (1230G>A) در اگزون یازدهم ژن SOS1 (تجزیه و تحلیل ژن PTPN11 - هیچ جهشی یافت نشد) شناسایی شد که مشخصه سندرم نونان است.
مشاوره تخصصی:
متخصص غدد- کم کاری تیروئید تحت بالینی، جبران ناقص دارویی.
چشم پزشک- آستیگماتیسم
متخصص مغز و اعصاب- دیستونی رویشی- عروقی. واکنش های عصبی
متخصص قلب- کاردیوپاتی عملکردی
جراح ارتوپد- وضعیت بدنی ضعیف بدشکلی قفسه سینه.
متخصص ژنتیک- سندرم نونان
با در نظر گرفتن فنوتیپ کودک، سابقه پزشکی و نتایج مطالعات تکمیلی، تشخیص سندرم نونان داده شد که نتیجه یک مطالعه ژنتیکی مولکولی تایید شد.
بنابراین، مشاهدات بالینی ارائه شده دشواری های یک جستجوی تشخیصی افتراقی، نیاز به ادغام علائم فردی در فنوتیپ کلی یک وضعیت پاتولوژیک خاص برای تشخیص به موقع و هدفمند اشکال فردی بیماری های ارثی، و اهمیت روش های ژنتیکی مولکولی برای روشن شدن را نشان می دهد. تشخیص تشخیص به موقع و روشن شدن پیدایش هر سندرم از اهمیت ویژه ای برخوردار است، زیرا به فرد امکان می دهد رویکرد بهینه برای درمان این شرایط و پیشگیری از عوارض احتمالی (تا و از جمله ناتوانی کودک) را پیدا کند. پیشگیری از عود بیماری های ارثی در خانواده های مبتلا (مشاوره پزشکی و ژنتیک). این امر نیاز به پزشکان تخصص های مختلف را برای هدایت واضح جریان آسیب شناسی ارثی تعیین می کند.
کتابشناسی - فهرست کتب:
- Baird P., De Jong B. Noonan's syndrome (فنوتیپ XX و XY Turner) در سه نسل از یک خانواده // J. Pediatr., 1972, vol. 80، ص. 110-114.
- Hasegawa T.، Ogata T. و همکاران. کوآرکتاسیون آئورت و هوپوپلازی کلیه در پسری با ناهنجاریهای سطحی ترنر/نونان و کاریوتیپ 46، XY: یک مدل بالینی برای اختلال احتمالی ژن(های) لنفوژنیک احتمالی برای کلالههای سوماتیک ترنر // هوم. ژنت.، 1996، ج. 97، ر. 564–567.
- Fedotova T.V.، Kadnikova V.A. و همکاران تجزیه و تحلیل ژنتیکی بالینی و مولکولی سندرم نونان. مواد کنگره ششم انجمن ژنتیک پزشکی روسیه. ژنتیک پزشکی، ضمیمه شماره 5، 1389، ص 184.
- Ward K.A., Moss C., McKeown C. سندرم قلبی-فاسیو-جلدی: تظاهری از سندرم نونان؟ // برادر J. Dermatol., 1994, vol. 131، ص. 270-274.
- مونیچی جی.، پاسکوینو A.M. و همکاران درمان هورمون رشد در سندرم نونان: گزارش چهار مورد که به قد نهایی رسیده اند // هورم. رس.، 1995، ج. 44، ر. 164-167.
فصل 5 تغییرپذیری ارگانیسم
فصل 5 تغییرپذیری ارگانیسم
اطلاعات کل
تنوع یک موجود زنده، تنوع ژنوم آن است که تفاوت های ژنوتیپی و فنوتیپی یک فرد را تعیین می کند و باعث تنوع تکاملی ژنوتیپ ها و فنوتیپ های آن می شود (به فصل های 2 و 3 مراجعه کنید).
رشد داخل رحمی جنین، جنین، جنین، رشد بیشتر پس از تولد بدن انسان (نوزادی، کودکی، نوجوانی، نوجوانی، بزرگسالی، پیری و مرگ) مطابق با برنامه ژنتیکی انتوژنز انجام می شود که از ادغام ژنوم های مادر و پدر (به فصل های 2 و 12 مراجعه کنید).
در طول آنتوژنز، ژنوم بدن یک فرد و اطلاعات رمزگذاری شده در آن تحت تأثیر عوامل محیطی تحت دگرگونی های مداوم قرار می گیرند. تغییراتی که در ژنوم رخ می دهد می تواند از نسلی به نسل دیگر منتقل شود و باعث تنوع در خصوصیات و فنوتیپ ارگانیسم در فرزندان می شود.
در آغاز قرن بیستم. جانورشناس آلمانی دبلیو هکر شاخه ای از ژنتیک را شناسایی کرد که به مطالعه پیوندها و روابط بین ژنوتیپ ها و فنوتیپ ها و تجزیه و تحلیل تنوع آنها اختصاص داشت و آن را نام برد. فنوژنتیک
در حال حاضر، فنوژنتیک ها دو دسته از تنوع را تشخیص می دهند: غیر ارثی (یا اصلاح شده)، که از نسلی به نسل دیگر منتقل نمی شود، و ارثی، که از نسلی به نسل دیگر منتقل می شود.
به نوبه خود، تنوع ارثی نیز به دو دسته تقسیم می شود: ترکیبی (بازترکیبی) و جهشی. تنوع طبقه اول با سه مکانیسم تعیین می شود: برخورد تصادفی گامت ها در طول لقاح. تقاطع یا نوترکیبی میوز (تبادل بخشهای مساوی بین کروموزومهای همولوگ در مرحله تقسیم اول میوز)؛ واگرایی مستقل کروموزوم های همولوگ به قطب های تقسیم در طول تشکیل سلول های دختر در طول میتوز و میوز. تغییرپذیری دوم
کلاس توسط جهش های نقطه ای، کروموزومی و ژنومی ایجاد می شود (به زیر مراجعه کنید).
اجازه دهید به طور متوالی طبقات و انواع تنوع ارگانیسم را در مراحل مختلف رشد فردی آن در نظر بگیریم.
تنوع در طی لقاح گامت ها و شروع عملکرد ژنوم ارگانیسم نوپا
ژنوم مادر و پدر نمی توانند جدا از یکدیگر عمل کنند.
تنها دو ژنوم والدین، که در یک زیگوت متحد شده اند، منشا حیات مولکولی، ظهور یک حالت کیفی جدید - یکی از ویژگی های ماده بیولوژیکی را فراهم می کنند.
در شکل شکل 23 نتایج برهمکنش دو ژنوم والدین را در طی لقاح گامت نشان می دهد.
با توجه به فرمول لقاح: زیگوت = تخمک + اسپرم، آغاز رشد زیگوت لحظه تشکیل یک دوتایی (دیپلوئید) است که دو مجموعه هاپلوئید از گامت های والدین به هم می رسند. پس از آن است که زندگی مولکولی به وجود می آید و زنجیره ای از واکنش های متوالی بر اساس بیان ژن های ژنوتیپ زیگوت و سپس بر روی ژنوتیپ های سلول های سوماتیک دختری که از آن بیرون آمده اند راه اندازی می شود. ژنها و گروههایی از ژنها در ژنوتیپهای تمام سلولهای بدن در طول اجرای برنامه ژنتیکی انتوژنز شروع به "روشن شدن" و "خاموش شدن" میکنند.
نقش اصلی در وقایعی که رخ می دهد متعلق به تخم است که در هسته و سیتوپلاسم همه چیز لازم برای جوانه زنی را دارد.
برنج. 23.نتایج تعامل دو ژنوم والدین در طول لقاح گامت (تصاویر از www.bio.1september.ru؛ www.bio.fizteh.ru؛ www. vetfac.nsau.edu.ru، به ترتیب)
توسعه و ادامه حیات، اجزای ساختاری و عملکردی هسته و سیتوپلاسم (جوهر) مادرسالاری بیولوژیکی).اسپرم حاوی DNA است و حاوی اجزای سیتوپلاسمی نیست. پس از نفوذ به تخمک، DNA اسپرم با DNA آن در تماس است و بنابراین مکانیسم مولکولی اصلی که در طول زندگی ارگانیسم عمل می کند در زیگوت "روشن" می شود: تعامل DNA-DNA دو ژنوم والدین. به طور دقیق، ژنوتیپ فعال می شود، که توسط قسمت های تقریبا مساوی از توالی های نوکلئوتیدی DNA با منشاء مادری و پدری (بدون در نظر گرفتن mtDNA سیتوپلاسم) نشان داده می شود. بیایید آنچه گفته شد ساده کنیم: شروع زندگی مولکولی در زیگوت نقض ثبات محیط داخلی تخم مرغ (هموستاز آن) است و کل زندگی مولکولی بعدی یک ارگانیسم چند سلولی تمایل به بازگرداندن هموستاز است. در معرض عوامل محیطی یا تعادل بین دو حالت متضاد: ثبات از یک طرفو تنوع با یکی دیگر.اینها روابط علت و معلولی هستند که ظهور و تداوم حیات مولکولی یک موجود زنده را در طول انتوژنز تعیین می کنند.
حال اجازه دهید به نتایج و اهمیت تنوع ژنوم یک موجود زنده به عنوان محصول تکامل توجه کنیم. ابتدا بیایید این سوال را در مورد منحصر به فرد بودن ژنوتیپ زیگوت یا سلول پیش ساز تمام سلول ها، بافت ها، اندام ها و سیستم های بدن در نظر بگیریم.
لقاح به خودی خود اتفاقی رخ می دهد: یک گامت ماده تنها توسط یک گامت نر از 200 تا 300 میلیون اسپرم موجود در انزال مرد بارور می شود. بدیهی است که هر تخمک و هر اسپرم با بسیاری از ویژگی های ژنوتیپی و فنوتیپی از یکدیگر متمایز می شوند: وجود ژن های تغییر یافته یا بدون تغییر در ترکیب و ترکیبات (نتایج تنوع ترکیبی)، توالی های مختلف توالی های نوکلئوتیدی DNA، اندازه ها، شکل های مختلف. ، فعالیت عملکردی (تحرک)، بلوغ گامت ها و غیره. این تفاوت ها هستند که به ما اجازه می دهند در مورد منحصر به فرد بودن ژنوم هر گامت و در نتیجه ژنوتیپ زیگوت و کل ارگانیسم صحبت کنیم: تصادف لقاح گامت ها تولد یک ارگانیسم منحصر به فرد ژنتیکی را تضمین می کنند.
به عبارت دیگر، زندگی مولکولی یک فرد (مثل زندگی یک موجود بیولوژیکی به طور کلی) یک "هدیه سرنوشت" یا اگر بخواهید "هدیه الهی" است، زیرا به جای یک فرد معین با همان
این احتمال وجود داشت که برادران و خواهران ژنتیکی متفاوتی به دنیا بیایند.
حال اجازه دهید بحث خود را در مورد تعادل بین ثبات و تنوع مواد ارثی ادامه دهیم. در یک مفهوم گسترده، حفظ چنین تعادلی حفظ و تغییر (تبدیل) همزمان پایداری مواد ارثی تحت تأثیر عوامل محیطی داخلی (هوموستاز) و خارجی (هنجار واکنش) است. هموستاز به ژنوتیپ ناشی از همجوشی دو ژنوم بستگی دارد (شکل 23 را ببینید). سرعت واکنش توسط برهمکنش ژنوتیپ با عوامل محیطی تعیین می شود.
هنجار و دامنه واکنش
روش خاص واکنش بدن در پاسخ به عوامل محیطی نامیده می شود هنجار واکنشاین ژن ها و ژنوتیپ هستند که مسئول توسعه و دامنه تغییرات خصوصیات فردی و فنوتیپ کل ارگانیسم هستند. در عین حال، تمام قابلیت های ژنوتیپ در فنوتیپ تحقق نمی یابد، یعنی. فنوتیپ یک مورد خاص (برای یک فرد) از اجرای یک ژنوتیپ در شرایط محیطی خاص است. بنابراین، به عنوان مثال، بین دوقلوهای تک تخمکی که دارای ژنوتیپ های کاملاً یکسان هستند (100٪ ژن مشترک)، اگر دوقلوها در شرایط محیطی مختلف رشد کنند، تفاوت های فنوتیپی قابل توجهی آشکار می شود.
هنجار واکنش می تواند محدود یا گسترده باشد. در حالت اول، ثبات یک صفت فردی (فنوتیپ) تقریباً بدون توجه به تأثیرات محیطی حفظ می شود. نمونه هایی از ژن ها با هنجار واکنش باریک یا ژن های غیر پلاستیکیژنهایی هستند که سنتز آنتیژنهای گروه خونی، رنگ چشم، فر کردن مو و غیره را کد میکنند. عملکرد آنها در هر شرایط خارجی (سازگار با زندگی) یکسان است. در حالت دوم، ثبات یک صفت فردی (فنوتیپ) بسته به تأثیر محیط تغییر می کند. نمونه ای از ژن هایی با سرعت واکنش گسترده یا ژن های پلاستیکی- ژن هایی که تعداد گلبول های قرمز خون را کنترل می کنند (برای افرادی که از کوه بالا می روند و افرادی که از کوه پایین می روند متفاوت است). نمونه دیگری از هنجار واکنش گسترده، تغییر رنگ پوست (برنزه شدن) است که با شدت و زمان قرار گرفتن در معرض اشعه ماوراء بنفش در بدن همراه است.
صحبت کردن در مورد محدوده پاسخ،باید تفاوت های فنوتیپی که در یک فرد (ژنوتیپ او) ظاهر می شود را در نظر داشت.
شرایط محیطی "تهی شده" یا "غنی" که ارگانیسم در آن قرار دارد. طبق تعریف I.I. Schmalhausen (1946)، "این ویژگی ها به این شکل نیست که به ارث می رسد، بلکه هنجار واکنش آنها به تغییرات در شرایط وجود موجودات است."
بنابراین، هنجار و دامنه واکنش، حدود تنوع ژنوتیپی و فنوتیپی ارگانیسم در هنگام تغییر شرایط محیطی است.
همچنین باید توجه داشت که از میان عوامل درونی مؤثر بر تظاهرات فنوتیپی ژن ها و ژنوتیپ، جنسیت و سن فرد از اهمیت خاصی برخوردار است.
عوامل بیرونی و درونی تعیین کننده رشد صفات و فنوتیپ ها در سه گروه از عوامل اصلی ذکر شده در فصل شامل ژن ها و ژنوتیپ، مکانیسم های تعاملات بین مولکولی (DNA-DNA) و بین ژنی بین ژنوم های والدین و عوامل محیطی قرار می گیرند.
البته اساس سازگاری ارگانیسم با شرایط محیطی (مبنای انتوژنز) ژنوتیپ آن است. به طور خاص، افراد با ژنوتیپ هایی که اثرات منفی ژن های پاتولوژیک و عوامل محیطی را سرکوب نمی کنند، نسبت به افرادی که اثرات نامطلوب در آنها سرکوب شده است، فرزندان کمتری بر جای می گذارند.
این احتمال وجود دارد که ژنوتیپهای موجودات زندهتر شامل ژنهای ویژه (ژنهای اصلاحکننده) باشد که عملکرد ژنهای «مضر» را بهگونهای سرکوب میکنند که به جای آن، آللهای نوع طبیعی غالب شوند.
تغییرپذیری غیر ارثی
در مورد تنوع غیر ارثی مواد ژنتیکی، اجازه دهید دوباره نمونه ای از یک هنجار واکنش گسترده را در نظر بگیریم - تغییر رنگ پوست تحت تأثیر اشعه ماوراء بنفش. "تن" از نسلی به نسل دیگر منتقل نمی شود، یعنی. ارثی نیست، اگرچه ژن های پلاستیکی در بروز آن دخیل هستند.
به همین ترتیب، نتایج صدمات، تغییرات اسکار در بافت ها و غشاهای مخاطی ناشی از بیماری سوختگی، سرمازدگی، مسمومیت و بسیاری از علائم دیگر که صرفاً توسط عوامل محیطی ایجاد می شود، ارثی نمی باشد. در عین حال باید تاکید کرد: تغییرات یا اصلاحات غیر ارثی با ارثی همراه است
خواص طبیعی یک موجود زنده، زیرا آنها در برابر پس زمینه یک ژنوتیپ خاص در شرایط محیطی خاص تشکیل می شوند.
تنوع ترکیبی ارثی
همانطور که در ابتدای فصل بیان شد، علاوه بر مکانیسم برخورد تصادفی گامت ها در حین لقاح، تنوع ترکیبی شامل مکانیسم های عبور در اولین تقسیم میوز و واگرایی مستقل کروموزوم ها به قطب های تقسیم در طول تشکیل دختر است. سلول ها در طول میتوز و میوز (به فصل 9 مراجعه کنید).
عبور در اولین تقسیم میوز
با توجه به مکانیسم عبور از رویپیوند ژن ها با کروموزوم به طور منظم در مرحله تقسیم اول میوز در نتیجه اختلاط (تبادل) ژن های منشاء پدری و مادری مختل می شود (شکل 24).
در آغاز قرن بیستم. هنگام باز کردن گذرگاه بر روی T.H. مورگان و شاگردانش پیشنهاد کردند که تلاقی بین دو ژن نه تنها در یک، بلکه در دو، سه (به ترتیب تقاطع دو و سه گانه) و بیشتر نیز می تواند رخ دهد. سرکوب عبور و مرور در مناطق بلافاصله در مجاورت نقاط مبادله مشاهده شد. این سرکوب نامیده شد دخالت.
در نهایت، محاسبه شد: برای یک میوز مرد از 39 تا 64 کیاسما یا نوترکیبی و برای یک میوز ماده تا 100 کیاسما وجود دارد.
 برنج. 24.طرح تقاطع در بخش اول میوز (طبق گفته شوچنکو V.A. و همکاران، 2004):
برنج. 24.طرح تقاطع در بخش اول میوز (طبق گفته شوچنکو V.A. و همکاران، 2004):
الف - کروماتیدهای خواهر کروموزوم های همولوگ قبل از شروع میوز. ب - آنها در طول پاکیتن هستند (مارپیچی شدن آنها قابل مشاهده است). ج - آنها همچنین در حین دیپلوتن و دیاکینزیس هستند (فلش ها مکان های عبور از روی کیاسما یا مناطق مبادله را نشان می دهند)
در نتیجه، آنها به این نتیجه رسیدند: پیوند ژن ها با کروموزوم ها به طور مداوم در حین عبور از یکدیگر مختل می شود.
عوامل موثر در عبور از گذر
کراس اوور یکی از فرآیندهای ژنتیکی منظم در بدن است که توسط بسیاری از ژن ها هم به طور مستقیم و هم از طریق وضعیت فیزیولوژیکی سلول ها در طول میوز و حتی میتوز کنترل می شود.
عوامل موثر بر عبور از مرز عبارتند از:
جنسیت همسان و هتروگامتیک (ما در مورد آن صحبت می کنیم عبور از میتوزدر نر و ماده یوکاریوت هایی مانند مگس سرکه و کرم ابریشم). بنابراین، در مگس سرکه عبور از آن به طور معمول انجام می شود. در کرم ابریشم یا طبیعی است یا وجود ندارد. در انسان، باید به جنسیت مختلط ("سوم") و به طور خاص به نقش تلاقی در ناهنجاری های رشد جنسی در هرمافرودیتیسم نر و ماده توجه شود (به فصل 16 مراجعه کنید).
ساختار کروماتین؛ فراوانی تلاقی در مناطق مختلف کروموزوم ها تحت تأثیر توزیع هتروکروماتیک (مناطق پرسانترومریک و تلومر) و نواحی یوکروماتیک است. به طور خاص، در مناطق پریسانترومری و تلومر، فرکانس عبور کاهش مییابد و فاصله بین ژنهایی که با فرکانس عبور تعیین میشود ممکن است با واقعی مطابقت نداشته باشد.
وضعیت عملکردی بدن؛ با افزایش سن، درجه مارپیچی شدن کروموزوم و سرعت تقسیم سلولی تغییر می کند.
ژنوتیپ؛ حاوی ژن هایی است که دفعات عبور را افزایش یا کاهش می دهد. "قفل های" دومی بازآرایی های کروموزومی (وارونگی و جابجایی) هستند که ترکیب طبیعی کروموزوم ها را در زیگوتن پیچیده می کنند.
عوامل برون زا: قرار گرفتن در معرض دما، پرتوهای یونیزان و محلول های نمک غلیظ، جهش زاهای شیمیایی، داروها و هورمون ها که معمولاً دفعات عبور را افزایش می دهند.
فرکانس تلاقی میوز و میتوزی و SCO گاهی اوقات برای قضاوت در مورد اثر جهش زایی داروها، مواد سرطان زا، آنتی بیوتیک ها و سایر ترکیبات شیمیایی استفاده می شود.
عبور نابرابر
در موارد نادر، هنگام عبور از کروماتیدها، شکستگی در نقاط نامتقارن کروماتیدهای خواهر مشاهده می شود و آنها تبادل می کنند.
بین خود به مناطق نابرابر تقسیم می شوند - این است عبور نابرابر
در عین حال، مواردی توصیف شده است که در طی میتوز، کونژوگه میتوزی (جفت شدن نادرست) کروموزوم های همولوگ مشاهده می شود و نوترکیبی بین کروماتیدهای غیر خواهر رخ می دهد. این پدیده نامیده می شود تبدیل ژن
اهميت اين مكانيزم به سختي قابل برآورد است. به عنوان مثال، در نتیجه جفت شدن نادرست کروموزوم های همولوگ در امتداد تکرارهای کناری، ممکن است دو برابر شدن (تکثیر) یا از دست دادن (حذف) ناحیه کروموزوم حاوی ژن PMP22 رخ دهد که منجر به ایجاد حسی حرکتی اتوزومال غالب ارثی می شود. نوروپاتی Charcot-Marie-Tooth.
عبور نابرابر یکی از مکانیسم های بروز جهش است. به عنوان مثال، پروتئین محیطی میلین توسط ژن PMP22 که در کروموزوم 17 قرار دارد و طولی در حدود 1.5 میلیون جفت باز دارد کدگذاری می شود. این ژن توسط دو تکرار همولوگ به طول تقریبی 30 کیلوبایت احاطه شده است. (تکرارها در کناره های ژن قرار دارند).
به خصوص بسیاری از جهش ها در نتیجه تلاقی نابرابر در شبه زا رخ می دهد. سپس یا قطعه ای از یک آلل به آلل دیگر منتقل می شود یا قطعه ای از یک شبه ژن به یک ژن منتقل می شود. به عنوان مثال، هنگامی که یک توالی شبه کاذب به ژن 21 هیدروکسیلاز (CYP21B) در سندرم آدرنوژنیتال یا هیپرپلازی مادرزادی آدرنال منتقل می شود، جهش مشابهی مشاهده می شود (فصل 14 و 22 را ببینید).
علاوه بر این، به دلیل نوترکیبی ها در طول تلاقی نابرابر، می توان اشکال آللی متعددی از ژن های کد کننده آنتی ژن های کلاس I HLA را تشکیل داد.
واگرایی مستقل کروموزوم های همولوگ به قطب های تقسیم در طول تشکیل سلول های دختر در طول میتوز و میوز
با توجه به فرآیند همانندسازی که قبل از میتوز یک سلول سوماتیک انجام می شود، تعداد کل توالی های نوکلئوتیدی DNA دو برابر می شود. تشکیل یک جفت کروموزوم همولوگ از دو کروموزوم پدری و دو کروموزوم مادری اتفاق می افتد. هنگامی که این چهار کروموزوم در دو سلول دختر توزیع می شوند، هر سلول یک کروموزوم پدری و یک کروموزوم مادری (برای هر جفت مجموعه کروموزوم) دریافت می کند، اما کدام یک از این دو، اولی یا دومی، ناشناخته است. رخ می دهد
توزیع تصادفی کروموزوم های همولوگ محاسبه آسان است: به دلیل ترکیبات مختلف 23 جفت کروموزوم، تعداد کل سلول های دختر 223 یا بیش از 8 میلیون (8 χ 10 6) گونه از ترکیب کروموزوم ها و ژن های واقع در آنها خواهد بود. در نتیجه، با توزیع تصادفی کروموزوم ها در سلول های دختر، هر یک از آنها کاریوتیپ و ژنوتیپ منحصر به فرد خود را خواهند داشت (نسخه خاص خود از ترکیب کروموزوم ها و ژن های مرتبط با آنها). لازم به ذکر است که یک نوع پاتولوژیک از توزیع کروموزوم ها در سلول های دختر وجود دارد. به عنوان مثال، ورود به یکی از دو سلول دختر تنها یک کروموزوم X (منشا پدری یا مادری) منجر به مونوزومی (سندرم شرشفسکی-ترنر، کاریوتایپ 45، XO) می شود، ورود سه اتوزوم یکسان منجر به تریزومی (داون) می شود. سندرم، 47، XY، + 21؛ Patau، 47، XX، + 13 و Edvadsa، 47، XX، + 18؛ همچنین به فصل 2 مراجعه کنید).
همانطور که در فصل 5 اشاره شد، دو کروموزوم پدری یا دو کروموزوم مادری می توانند به طور همزمان وارد یک سلول دختر شوند - این ایزودیزومی تک والدینی برای یک جفت کروموزوم خاص است: سندرم سیلور-راسل (دو کروموزوم مادری 7)، بک ویت-ویدمن (سندرم پدری). کروموزوم 11)، آنجلمن (دو کروموزوم پدری 15)، پرادر-ویلی (دو کروموزوم مادری 15). به طور کلی حجم اختلالات توزیع کروموزوم به 1 درصد کل اختلالات کروموزومی در انسان می رسد. این اختلالات از نظر تکاملی اهمیت زیادی دارند، زیرا تنوع جمعیتی کاریوتیپها، ژنوتیپها و فنوتیپهای انسانی را ایجاد میکنند. علاوه بر این، هر گونه آسیب شناسی محصول منحصر به فرد تکامل است.
در نتیجه تقسیم دوم میوز، 4 سلول دختر تشکیل می شود. هر کدام از آنها یک کروموزوم مادری یا پدری را از هر 23 کروموزوم دریافت خواهند کرد.
برای جلوگیری از خطاهای احتمالی در محاسبات بعدی، آن را به عنوان یک قاعده در نظر می گیریم: در نتیجه تقسیم دوم میوز، 8 میلیون نوع گامت نر و 8 میلیون نوع گامت ماده نیز تشکیل می شود. سپس پاسخ به این سوال که حجم کل ترکیبات واریانت کروموزوم ها و ژن هایی که روی آنها قرار می گیرند در هنگام برخورد دو گامت چقدر است، به شرح زیر است: 2 46 یا 64 χ 10 12، یعنی. 64 تریلیون
تشکیل چنین تعداد ژنوتیپ (از لحاظ نظری ممکن) هنگام ملاقات دو گامت به وضوح معنای ناهمگونی ژنوتیپ ها را توضیح می دهد.
ارزش تنوع ترکیبی
تنوع ترکیبی نه تنها برای ناهمگنی و منحصر به فرد بودن مواد ارثی، بلکه برای بازیابی (ترمیم) پایداری مولکول DNA در زمانی که هر دو رشته آسیب می بینند، مهم است. به عنوان مثال، ایجاد شکاف DNA تک رشته ای در مقابل یک ضایعه ترمیم نشده است. شکاف حاصل را نمی توان بدون درگیر کردن رشته DNA طبیعی در ترمیم به دقت اصلاح کرد.
تنوع جهشی
همراه با منحصر به فرد بودن و ناهمگونی ژنوتیپ ها و فنوتیپ ها در نتیجه تنوع ترکیبی، سهم بزرگی در تغییرپذیری ژنوم و پدیده انسان توسط تنوع جهش ارثی و ناهمگنی ژنتیکی حاصل می شود.
تغییرات در توالی های نوکلئوتیدی DNA را می توان به طور معمول به جهش و پلی مورفیسم ژنتیکی تقسیم کرد (به فصل 2 مراجعه کنید). در عین حال، اگر ناهمگنی ژنوتیپ ها مشخصه های ثابت (طبیعی) تنوع ژنوم باشد، آنگاه تنوع جهشی- این معمولاً آسیب شناسی آن است.
تنوع پاتولوژیک ژنوم، به عنوان مثال، با عبور نابرابر، واگرایی نادرست کروموزوم ها به قطب های تقسیم در طول تشکیل سلول های دختر، وجود ترکیبات ژنتیکی و سری های آللی پشتیبانی می شود. به عبارت دیگر، تنوع ترکیبی و جهشی ارثی در انسان با تنوع ژنوتیپی و فنوتیپی قابل توجهی آشکار می شود.
اجازه دهید اصطلاحات را روشن کنیم و مسائل کلی نظریه جهش را در نظر بگیریم.
مسائل کلی در نظریه جهش ها
جهشتغییری در سازماندهی ساختاری، کمیت و/یا عملکرد مواد ارثی و پروتئین های سنتز شده توسط آن وجود دارد. این مفهوم اولین بار توسط Hugo de Vries ارائه شد
در 1901-1903 در کار خود "تئوری جهش"، جایی که او ویژگی های اساسی جهش ها را توضیح داد. آنها:
ناگهان ظاهر شود؛
از نسلی به نسل دیگر منتقل می شود؛
بر اساس نوع غالب (که در هتروزیگوت ها و هموزیگوت ها ظاهر می شود) و نوع مغلوب (در هموزیگوت ها آشکار می شود) به ارث می رسد.
آنها هیچ جهتی ندارند ("جهش" هر مکان، ایجاد تغییرات جزئی یا تاثیر بر علائم حیاتی).
با توجه به تظاهرات فنوتیپی آنها، آنها می توانند مضر (بیشتر جهش ها)، مفید (بسیار نادر) یا بی تفاوت باشند.
در سلول های سوماتیک و زایا رخ می دهد.
علاوه بر این، جهش های مشابه می تواند به طور مکرر رخ دهد.
فرآیند جهشیا جهش زایی، یک فرآیند مداوم در حال شکل گیری جهش ها تحت تأثیر عوامل جهش زا است - عوامل محیطی که به مواد ارثی آسیب می رساند.
اولین نظریه جهش زایی پیوستهدر سال 1889 توسط دانشمند روسی از دانشگاه سن پترزبورگ S.I. کورژینسکی در کتاب خود "ناهمگنی و تکامل".
همانطور که در حال حاضر اعتقاد بر این است، جهش ها می توانند به طور خود به خود، بدون علل خارجی قابل مشاهده ظاهر شوند، اما تحت تأثیر شرایط داخلی سلول و بدن - اینها جهش های خود به خود یا جهش زایی خود به خود
جهش هایی که به طور مصنوعی در اثر قرار گرفتن در معرض عوامل خارجی با ماهیت فیزیکی، شیمیایی یا بیولوژیکی ایجاد می شوند، جهش های القایی هستند، یا جهش زایی ناشی از
شایع ترین جهش ها نامیده می شوند جهش های عمده(به عنوان مثال، جهش در ژن های دیستروفی عضلانی دوشن بکر، فیبروز کیستیک، کم خونی سلول داسی شکل، فنیل کتونوری و غیره). اکنون کیت های تجاری ساخته شده اند که شناسایی خودکار مهمترین آنها را ممکن می سازد.
جهش های تازه ایجاد شده را جهش یا جهش جدید می نامند از نوبرای مثال، این جهشها شامل جهشهایی هستند که زمینهساز تعدادی از بیماریهای اتوزومال غالب هستند، مانند آکندروپلازی (10٪ موارد بیماری اشکال خانوادگی هستند)، نوروفیبروماتوز Recklinghausen نوع I (50-70٪ اشکال خانوادگی هستند)، بیماری آلزایمر، کره هانتینگتون. .
جهش از حالت طبیعی یک ژن (ویژگی) به حالت پاتولوژیک نامیده می شود سر راست.
جهش از حالت پاتولوژیک یک ژن (ویژگی) به حالت طبیعی معکوس یا نامیده می شود برگشت ها
توانایی بازگشت اولین بار در سال 1935 توسط N.V. تیموفیف-رسوفسکی.
جهش های بعدی در ژن که فنوتیپ جهش یافته اولیه را سرکوب می کند نامیده می شود. سرکوب کنندهسرکوب ممکن است باشد درون ژنی(فعالیت عملکردی پروتئین را بازیابی می کند؛ اسید آمینه با اسید آمینه اصلی مطابقت ندارد، یعنی برگشت پذیری واقعی وجود ندارد) و غیر ژنتیکی(ساختار tRNA تغییر می کند، در نتیجه tRNA جهش یافته به جای اسید آمینه ای که توسط سه گانه معیوب کدگذاری می شود، اسید آمینه دیگری را در پلی پپتید شامل می شود).
جهش در سلول های سوماتیک نامیده می شود جهش های جسمیآنها کلون های سلولی پاتولوژیک (مجموعه ای از سلول های پاتولوژیک) را تشکیل می دهند و در صورت حضور همزمان سلول های طبیعی و پاتولوژیک در بدن، منجر به موزائیسم سلولی می شوند (به عنوان مثال، در استئودیستروفی ارثی آلبرایت، بیان بیماری بستگی به تعداد سلول های غیر طبیعی).
جهش های سوماتیک می توانند خانوادگی یا پراکنده (غیر فامیلی) باشند. آنها زمینه ساز توسعه نئوپلاسم های بدخیم و فرآیندهای پیری زودرس هستند.
پیش از این، این یک اصل بدیهی در نظر گرفته می شد که جهش های جسمی ارثی نیستند. در سال های اخیر، انتقال استعداد ارثی از نسلی به نسل دیگر 90 درصد از اشکال چند عاملی و 10 درصد از اشکال تک ژنی سرطان، که با جهش در سلول های سوماتیک آشکار می شود، به اثبات رسیده است.
جهش در سلول های زایا نامیده می شود جهش های ژرمینالاعتقاد بر این است که آنها کمتر از جهش های جسمی شایع هستند، زمینه ساز همه بیماری های ارثی و مادرزادی هستند، از نسلی به نسل دیگر منتقل می شوند و همچنین می توانند خانوادگی یا پراکنده باشند. بیشترین منطقه مورد مطالعه جهش زایی عمومی، فیزیکی و به ویژه، جهش زایی تشعشعهر منبع پرتوهای یونیزان برای سلامتی انسان مضر است، آنها، به عنوان یک قاعده، دارای یک اثر قوی جهش زا، تراتوژن و سرطان زا هستند. اثر جهش زایی یک دوز پرتو بسیار بیشتر از پرتوهای مزمن است. دوز تابش 10 راد میزان جهش را در انسان دو برابر می کند. ثابت شده است که تشعشعات یونیزان می توانند جهش هایی ایجاد کنند که منجر به جهش می شود
به بیماری های ارثی (مادرزادی) و انکولوژیک و اشعه ماوراء بنفش - برای القای خطاهای تکرار DNA.
بزرگترین خطر است جهش زایی شیمیاییحدود 7 میلیون ترکیب شیمیایی در جهان وجود دارد. تقریباً 50-60 هزار ماده شیمیایی به طور مداوم در اقتصاد ملی، در تولید و در زندگی روزمره استفاده می شود. هر ساله حدود هزار ترکیب جدید وارد عمل می شود. از این تعداد 10 درصد قادر به ایجاد جهش هستند. اینها عبارتند از علف کش ها و آفت کش ها (سهم جهش زاها در میان آنها به 50٪ می رسد)، و همچنین تعدادی از داروها (برخی آنتی بیوتیک ها، هورمون های مصنوعی، سیتواستاتیک و غیره).
نیز وجود دارد جهش زایی بیولوژیکیجهش زاهای بیولوژیکی عبارتند از: پروتئین های خارجی واکسن ها و سرم ها، ویروس ها (واریسلا، سرخجه سرخجه، فلج اطفال، هرپس سیمپلکس، ایدز، آنسفالیت) و DNA، عوامل برون زا (تغذیه ضعیف پروتئین)، ترکیبات هیستامینی و مشتقات آن، هورمون های استروئیدی (عوامل درون زا) . تقویت اثر جهش زاهای خارجی comutagens(سموم).
تاریخ ژنتیک نمونه های زیادی از اهمیت ارتباط بین ژن ها و صفات دارد. یکی از آنها طبقه بندی جهش ها بسته به اثر فنوتیپی آنها است.
طبقه بندی جهش ها بسته به اثر فنوتیپی آنها
این طبقه بندی جهش ها برای اولین بار در سال 1932 توسط G. Möller ارائه شد. بر اساس طبقه بندی، موارد زیر شناسایی شدند:
جهش های آمورف این وضعیتی است که در آن صفت کنترل شده توسط آلل پاتولوژیک بیان نمی شود زیرا آلل پاتولوژیک در مقایسه با آلل طبیعی غیر فعال است. چنین جهش هایی شامل ژن آلبینیسم (11q14.1) و حدود 3000 بیماری اتوزومال مغلوب می شود.
جهش های آنتی مورفیک در این حالت، مقدار صفت کنترل شده توسط آلل پاتولوژیک در مقابل مقدار صفت کنترل شده توسط آلل نرمال است. چنین جهش هایی شامل ژن حدود 5-6 هزار بیماری اتوزومال غالب است.
جهش های هیپرمورفیک در مورد چنین جهشی، صفت کنترل شده توسط آلل پاتولوژیک بارزتر از صفت کنترل شده توسط آلل طبیعی است. مثال - gete-
حامل های روزیگوتیک ژن برای بیماری های ناپایداری ژنوم (به فصل 10 مراجعه کنید). تعداد آنها حدود 3٪ از جمعیت زمین (تقریبا 195 میلیون نفر) است و تعداد خود بیماری ها به 100 نوزولوژی می رسد. از جمله این بیماری ها: کم خونی فانکونی، آتاکسی تلانژکتازی، خشکی پوست، سندرم بلوم، سندرم های پروژروئید، بسیاری از انواع سرطان و غیره. و در خود بیماران (هموزیگوت های این ژن ها) بروز سرطان ده ها برابر بیشتر از حد طبیعی است.
جهش های هیپومورفیک این وضعیتی است که در آن بیان یک صفت کنترل شده توسط یک آلل پاتولوژیک در مقایسه با صفت کنترل شده توسط یک آلل طبیعی ضعیف می شود. چنین جهشهایی شامل جهش در ژنهای سنتز رنگدانه (1q31؛ 6p21.2؛ 7p15-q13؛ 8q12.1؛ 17p13.3؛ 17q25؛ 19q13؛ Xp21.2؛ Xp21.3؛ Xp22) و همچنین بیشتر از 00 بیماری های اتوزومال مغلوب
جهش های نئومورفیک گفته می شود که چنین جهشی زمانی رخ می دهد که صفت کنترل شده توسط آلل پاتولوژیک در مقایسه با صفت کنترل شده توسط آلل نرمال، کیفیت متفاوتی (جدید) داشته باشد. مثال: سنتز ایمونوگلوبولین های جدید در پاسخ به نفوذ آنتی ژن های خارجی به بدن.
در مورد اهمیت پایدار طبقهبندی G. Möller، لازم به ذکر است که 60 سال پس از انتشار، اثرات فنوتیپی جهشهای نقطهای بسته به تأثیری که بر ساختار محصول پروتئینی ژن داشتند به کلاسهای مختلفی تقسیم شدند. / یا سطح بیان آن.
به ویژه، برنده جایزه نوبل، ویکتور مک کوزیک (1992) جهش هایی را شناسایی کرد که توالی اسیدهای آمینه را در یک پروتئین تغییر می دهند. مشخص شد که آنها مسئول تظاهرات 50-60٪ موارد بیماری های تک ژنی هستند و جهش های باقی مانده (40-50٪ موارد) جهش های موثر بر بیان ژن را تشکیل می دهند.
تغییر در ترکیب اسید آمینه پروتئین خود را در یک فنوتیپ پاتولوژیک نشان می دهد، به عنوان مثال، در موارد متهموگلوبینمی یا کم خونی سلول داسی شکل ناشی از جهش در ژن بتاگلوبین. به نوبه خود، جهش های مؤثر بر بیان ژن طبیعی جدا شد. آنها منجر به تغییر در مقدار محصول ژن می شوند و با فنوتیپ های مرتبط با کمبود یک پروتئین خاص ظاهر می شوند، به عنوان مثال،
در موارد کم خونی همولیتیک،ناشی از جهش ژن های موضعی بر روی اتوزوم ها: 9q34.3 (کمبود آدنیلات کیناز). 12p13.1 (کمبود تریوسفسفات ایزومراز)؛ 21q22.2 (کمبود فسفوفروکتوکیناز).
طبقه بندی جهش ها توسط V. McKusick (1992) البته نسل جدیدی از طبقه بندی ها است. در همان زمان، در آستانه انتشار آن، طبقه بندی جهش ها بسته به سطح سازماندهی مواد ارثی به طور گسترده پذیرفته شد.
طبقه بندی جهش ها بسته به سطح سازماندهی مواد ارثی
طبقه بندی شامل موارد زیر است.
جهش های نقطه ای(نقض ساختار ژن در نقاط مختلف).
به بیان دقیق، جهشهای نقطهای شامل تغییرات در نوکلئوتیدها (پایههای) یک ژن است که منجر به تغییر در کمیت و کیفیت محصولات پروتئینی آنها میشود. تغییرات پایه عبارت است از جایگزینی، درج، حرکت یا حذف آنها، که می تواند با جهش در نواحی تنظیم کننده ژن ها (پرموتر، محل پلی آدنیلاسیون)، و همچنین در مناطق کد کننده و غیر کد کننده ژن ها (اگزون ها و اینترون ها، پیوند) توضیح داده شود. سایت های). جایگزینی پایه منجر به سه نوع کدون جهش یافته می شود: جهش های نادرست، جهش های خنثی و جهش های بی معنی.
جهش های نقطه ای به عنوان صفات ساده مندلی به ارث می رسند. آنها شایع هستند: 1 مورد در 200-2000 تولد - هموکروماتوز اولیه، سرطان روده بزرگ غیر پولیپوز، سندرم مارتین بل و فیبروز کیستیک.
جهش های نقطه ای، که بسیار نادر هستند (1:1،500،000)، نقص ایمنی ترکیبی شدید (SCID) ناشی از کمبود آدنوزین دآمیناز هستند. گاهی اوقات جهش های نقطه ای نه به دلیل قرار گرفتن در معرض جهش زاها، بلکه به عنوان خطا در همانندسازی DNA ایجاد می شوند. علاوه بر این، فرکانس آنها از 1:10 5 -1:10 10 تجاوز نمی کند، زیرا آنها با کمک سیستم های تعمیر سلولی تقریباً توسط آنها اصلاح می شوند.
جهش های ساختارییا انحرافات کروموزومی (ساختار کروموزوم ها را مختل می کند و منجر به تشکیل گروه های پیوندی ژنی جدید می شود). اینها حذف (از دست دادن)، تکرار (دو برابر شدن)، جابجایی (حرکت)، وارونگی (چرخش 180 درجه) یا درج (درج) مواد ارثی هستند. چنین جهش هایی از ویژگی های جسمی هستند
سلول های منطقی (از جمله سلول های بنیادی). فرکانس آنها 1 در 1700 تقسیم سلولی است.
تعدادی از سندرم ها ناشی از جهش های ساختاری هستند. معروف ترین نمونه ها: سندرم "گریه گربه" (کاریوتیپ: 46,ХХ,5р-)، سندرم Wolf-Hirschhorn (46,ХХ,4р-), شکل انتقال سندرم داون (کاریوتیپ: 47, ХУ, t ( 14; 21)).
مثال دیگر سرطان خون است. هنگامی که آنها رخ می دهند، بیان ژن در نتیجه به اصطلاح جدایی (جابه جایی بین بخش ساختاری ژن و ناحیه پروموتر آن) مختل می شود و در نتیجه سنتز پروتئین مختل می شود.
ژنومیک(عددی) جهش ها- نقض تعداد کروموزوم ها یا قطعات آنها (با افزودن یا از دست دادن کل کروموزوم ها یا قطعات آنها منجر به ظهور ژنوم های جدید یا قطعات آنها می شود). منشأ این جهشها به دلیل عدم تفکیک کروموزوم در میتوز یا میوز است.
در مورد اول، اینها آنئوپلوئیدها، تتراپلوئیدها با سیتوپلاسم تقسیم نشده، پلی پلوئیدها با 6، 8، 10 جفت کروموزوم یا بیشتر هستند.
در مورد دوم، این جدا نشدن کروموزوم های زوجی است که در تشکیل گامت ها (مونوسومی، تریزومی) یا لقاح یک تخمک توسط دو اسپرم (دیسپرمی یا جنین تری پلوئید) نقش دارند.
نمونه های معمولی آنها قبلاً بیش از یک بار ذکر شده است - اینها عبارتند از سندرم Shereshevsky-Turner (45، XX)، سندرم Klinefelter (47، XXY)، تریزومی منظم در سندرم داون (47، XX، +21).
در F2 افراد با یک صفت غالب می توانند هموزیگوت (AA) باشند.
و هتروزیگوت (Aa) حامل آلل غالب. برای پیدا کردن، لازم است یک تلاقی تحلیلی از چنین فردی با هموزیگوت مغلوب انجام شود. اگر فرد مورد مطالعه یک هموزیگوت غالب باشد، آنگاه همه فرزندان حاصل از این تلاقی دارای یک صفت غالب و در عین حال هتروزیگوت (Aa) خواهند بود. در حالت دوم، فرزندان به احتمال یکسان دارای افرادی با هر دو صفت غالب (Aa) و مغلوب (aa) خواهند بود.
قبلاً گفتیم که روشهای آنالیز هیبریدولوژیک برای انسان قابل اجرا نیستند. تعیین نوع وراثت فقط بر اساس تجزیه و تحلیل شجره نامه انجام می شود. در برخی موارد اتوزومال
بیماری غالب در یکی از والدین بیمار وجود دارد. علاوه بر این، صرف نظر از جنسیت، احتمال بروز این صفت در فرزندان یک حامل هتروزیگوت یک جهش غالب 50٪ است و
هموزیگوت - 100٪. اما اغلب (تا 90٪ موارد) بیماری های غالب نتیجه یک جهش de novo هستند. در این حالت به صورت بیماری های پراکنده ظاهر می شوند.
با نوع توارث اتوزومال مغلوب، این صفت در هیبریدهای نسل اول وجود ندارد، اما در F2، احتمال تولد افراد با صفت مغلوب بدون توجه به جنسیت آنها 25٪ خواهد بود.
هنگام انجام یک تلاقی تجزیه و تحلیل، یک صفت مغلوب و همچنین یک ویژگی غالب در نیمی از فرزندان مشاهده می شود. افراد دارای صفت مغلوب حاملان هموزیگوت آلل مغلوب (aa) هستند. بیشتر اوقات، آنها در فرزندان والدین هتروزیگوت ظاهر می شوند که خود دارای صفت مغلوب نیستند، اما ناقلان هتروزیگوت جهش هستند. چنین والدینی نامیده می شوند
هتروزیگوت های اجباری طبق قانون مندل، احتمال داشتن فرزند بیمار در هتروزیگوت های اجباری 25 درصد است. اگر ما در مورد یک بیماری اتوزومال مغلوب صحبت می کنیم، والدین کودک مبتلا معمولا سالم هستند، اما ممکن است چندین فرزند مبتلا داشته باشند. کودکان با
یک بیماری اتوزومال مغلوب اغلب در ازدواج های فامیلی متولد می شود و احتمال داشتن فرزند مبتلا با میزان رابطه بین والدین افزایش می یابد. جهش های اتوزومال مغلوب می توانند در یک جمعیت تجمع کنند زیرا حامل های هتروزیگوت تحت فشار انتخاب نیستند. اگر والدین یک کودک بیمار با هم فامیلی نداشته باشند، اغلب آنها حامل جهش های مختلف در یک ژن هستند و
فرزندان مبتلا آنها هر یک از این جهش ها را به ارث می برند، یعنی هستند
هتروزیگوت های مرکب. یک نوع توارث اتوزومال مغلوب مشخصه اکثر آنزیموپاتی های ارثی است.
ویژگی های وراثت صفات توسط ژن ها تعیین می شود
موضعی در کروموزوم های جنسی با این واقعیت توضیح داده می شود که در Y-
ژن های کمی روی کروموزوم وجود دارد و عملاً هیچ همولوگ از ژن های کروموزوم X وجود ندارد.
در نتیجه، آلل های مغلوب اکثر ژن های کروموزوم X در مردان ظاهر می شود. این وضعیت یک آلل مغلوب،
هنگامی که فاقد همولوگ باشد - (a/-)، همیزیگوت نامیده میشود.
توجه داشته باشید که این اصطلاح نه تنها به ژن های موضعی در کروموزوم های جنسی اطلاق می شود، بلکه در مواردی که ناحیه محلی سازی این ژن در یکی از کروموزوم های همولوگ حذف شده است، یعنی وجود ندارد، به ژن های اتوزومال نیز اشاره دارد.
با وراثت وابسته به X، تفاوت های فنوتیپی در فرزندان بسته به جهت تلاقی مشاهده می شود، یعنی بسته به وجود یک صفت در مادر یا پدر در نسل والد. اگر این صفت در مادر هموزیگوت غالب و وجود داشته باشد،
سپس در F1 همه افراد بدون در نظر گرفتن جنسیت، این ویژگی را خواهند داشت و در F2 یک تقسیم 3:1 مشاهده می شود که این صفت تنها در نیمی از افراد مذکر وجود ندارد. در فرزندان مادر هتروزیگوت، احتمال به دنیا آوردن افرادی با صفت غالب 50 درصد خواهد بود.
صرف نظر از جنسیت. اگر صفت غالب در نسل والدین در پدر باشد، در نسل اول این ویژگی خواهد بود.
فقط در دختران وجود داشته باشد، و در دوم - در هر دو دختر و پسر با احتمال 50٪.
با یک نوع ارثی مغلوب وابسته به جنس، این صفت اغلب در افراد مذکر تشخیص داده می شود و انتقال این صفت از "پدربزرگ" به "نوه" مشاهده می شود. این بیماری هرگز از پدر به پسر منتقل نمی شود، زیرا پسر کروموزوم X پدر را به ارث نمی برد، این بیماری همیشه منشأ مادری دارد. در بیشتر موارد، مردانی که دارای صفت مغلوب وابسته به جنس هستند، 50 درصد احتمال دارد در فرزندان مادران هتروزیگوت ظاهر شوند که این ویژگی را ندارند. تمام فرزندان نسل اول پدری که دارای صفت مغلوب هستند، این ویژگی را ندارند، اما نیمی از دختران او این جهش را در حالت هتروزیگوت و احتمال به دنیا آوردن نرهایی با صفت مغلوب دارند، همانطور که قبلاً گفتیم. ، 50 درصد خواهد بود. نوع وراثت صفاتی که توسط ژن های کروموزوم Y تعیین می شود، holandric نامیده می شود و با
انتقال یک صفت از پدر به پسر
در دهه های اخیر، حقایق زیادی انباشته شده است که نشان می دهد
در مورد وجود تعداد زیادی انحراف از انواع مندلیف
وراثت. به بیماری های غیر مندلی با نوع غیر سنتی
وراثت شامل بیماریهای میتوکندری، ناهنجاریهای تک والدینی و بیماریهای منقوش ژنومی و همچنین بیماریهای گسترش میشود.
ناشی از وجود جهش های پویا نوع میتوکندری یا سیتوپلاسمی توارث مادری نامیده می شود.
سلول های زایای نر، اگرچه حاوی تعداد بسیار کمی میتوکندری هستند که تحرک آنها را تضمین می کند، اما آنها را به فرزندان خود منتقل نمی کنند. بنابراین، تمام میتوکندری های جنین، صرف نظر از جنسیت آن، دارند
پسران و دختران به طور یکسان در آینده، با استفاده از نمونه بیماری های مختلف انسان، با جزئیات بیشتری درباره انواع وراثت بحث خواهیم کرد.
اجازه دهید یک بار دیگر تأکید کنیم که الگوهای وراثت مورد بحث در بالا برای صفات تک ژنی معتبر هستند. در فهرست ژنهای انسانی و بیماریهای ژنتیکی که طی چند دهه گذشته با مشارکت مستقیم و راهنمایی ژنتیکدان برجسته پزشکی عصر ما، ویکتور مککازیک (McKusick V. A. Mendelian inheritance in man: a catalog of human) گردآوری شده است. ژن ها و اختلال ژنتیکی -2006 - http://www.ncbi.nlm.nih.gov /OMIM/)، شرحی از بیش از 16000 ژن مسئول صفات تک ژنی فنوتیپی ارائه شده است. نحوه توارث برای تقریباً 11000 نفر از آنها ایجاد شده است و بیش از 8000 ژن انسانی نقشه برداری شده است. نزدیک
4500 ژن با بیماری های تک ژنی مختلف مرتبط هستند. برای تقریباً 4000 بیماری تک ژنی، نحوه توارث تعیین شده است. تعداد بیماریهای اتوزومی بیش از 3500 است و تعداد بیماریهای غالب و مغلوب تقریباً یکسان است، اگرچه هنوز موارد غالب کمی بیشتر است. بیش از 300 بیماری به روش ایکس به ارث می رسند،
فقط تعداد کمی (نه بیشتر از 10) - پیوند Y و کمی بیشتر از 20
بیماری ها در اثر جهش در ژن های میتوکندری ایجاد می شوند.
در برخی موارد، هیچ یک از والدین ناقل جهش موجود در فرزندشان نیستند. ما قبلاً نوشتهایم که جهشها در یک ژن خاص میتوانند در طول گامتوژنز در یکی از سلولهای زایای والدین بهطور ناگهانی ایجاد شوند. برخی از بیماری های اتوزومال غالب به طور کامل به دلیل جهش های de novo هستند. اینها شامل آکندروپلازی است که در آن اکثر بیماران دارای یک جهش خاص در ژن گیرنده فاکتور رشد فیبروبلاست 3 (Fgf) هستند. تقریباً همه موارد به صورت de novo رخ می دهند
جایگزینی یکی از اسیدهای آمینه همولوگ (پرولین) در ژن های سه Fgf-
گیرنده های شناسایی شده در بیماران مبتلا به انواع ارثی کرانیوسینوستوز (همجوشی نامنظم بخیه های جمجمه در
کودک). فرکانس این جهش ها سه مرتبه بزرگتر از حد معمول است. مکان های وقوع این جهش های خاص جزو قابل تغییر ترین مکان ها در ژنوم انسان هستند یا گفته می شود.
"نقاط داغ" جهش زایی ، و دلایل این بی ثباتی بسیار خاص هنوز ناشناخته است.
فراوانی جهش در ژن های دیستروفی عضلانی دوشن و هموفیلی A افزایش یافته است.تقریباً 40-45 درصد از بیماران مبتلا به این بیماری های مرتبط با X دارای جهش های نو هستند. هنگام ارائه مشاوره ژنتیک پزشکی به چنین بیمارانی، بسیار مهم است که مشخص شود آیا بیمار این جهش را از مادر هتروزیگوت خود به ارث برده است یا اینکه آیا این جهش به صورت de novo ایجاد شده است.
در مورد اول، در چنین خانواده ای با حاملگی های مکرر، لازم است اقدامات پیشگیرانه خاصی با هدف جلوگیری از تولد یک کودک بیمار انجام شود. در حالت دوم، خطر تولد مجدد یک کودک بیمار در یک خانواده معین از ارزش عمومی جمعیت فراتر نمی رود و این خانواده نیازی به اقدامات پیشگیرانه ندارد. بعداً در مورد این وضعیت با جزئیات بیشتر صحبت خواهیم کرد.
بیش از یک ژن ممکن است در کنترل ژنتیکی اکثریت قریب به اتفاق صفات موجود در یک موجود زنده نقش داشته باشد. در این مورد آنها صحبت می کنند
وراثت چند ژنی گاهی اوقات تعداد این ژن ها به ده ها یا حتی صدها می رسد. وراثت چند ژنی معمولی است، به ویژه،
برای صفات کمی که شاخص های آن قابل اندازه گیری است، مانند قد، وزن، امید به زندگی و بسیاری از خواص تولیدی گیاهان و حیوانات کشاورزی. تنوع در تظاهرات فنوتیپی چنین صفاتی در جمعیت ها با توزیع نرمال مطابقت دارد - شکل. 12.
شکل 12. نمونه ای از توزیع نرمال
کلاس صفات ارثی چند ژنی شامل بسیاری از بیماری های انسانی گسترده است - آترواسکلروز، فشار خون بالا، دیابت شیرین، بیماری زخم معده، آسم برونش و بسیاری از بیماری های چند عاملی دیگر. برای مطالعه وراثت کمی و سایر صفات چند ژنی، از روش های آماری توسعه یافته در نیمه اول قرن گذشته توسط فیشر استفاده می شود.
فصل 1.6. ژنتیک جمعیت
هر نوع ارگانیسم با سطح معینی از تنوع ژنوتیپی مشخص می شود که ماهیت آن در جمعیت های مختلف متفاوت است. بررسی تنوع ژنتیکی جمعیت ها و الگوهای نگهداری از آن موضوع علم جمعیت است
ژنتیک پایه و اساس توسعه این حوزه از ژنتیک، کار S. S. Chetverikov "در برخی از جنبه های روند تکاملی از دیدگاه ژنتیک مدرن" بود که در سال 1926 منتشر شد. برای اولین بار در مورد مسائل حفظ جهش در جمعیت های طبیعی بحث می کند،
تأثیر بر این فرآیند انتخاب و انزوا و همچنین اهمیت آنها در تکامل.
در جمعیت های بزرگی که در آنها ترجیحی در تشکیل زوج های متاهل بر اساس ویژگی های مرتبط، ملی، مذهبی، اجتماعی یا سایر ویژگی ها وجود ندارد (این گونه جمعیت ها از کلمه پانمیکسیا - تلاقی تصادفی نامیده می شوند)، ارتباط بین فراوانی آلل ها و ژنوتیپ ها مطابق با قانون هاردی واینبرگ است که به طور مستقل توسط این دو دانشمند در سال 1908 کشف شد. برای صفات تک ژنی، به نظر می رسد: اگر فراوانی آلل های A ia برابر با p و q باشد، فرکانس هموزیگوت های AA iaa برابر با p2 و q2 و هتروزیگوت ها Aa - 2pq خواهد بود.
به ترتیب.
با انتخاب علیه یک طبقه ژنوتیپی خاص، جهش یا همخونی، که در ازدواج های نزدیک و در جمعیت های کوچک جدا شده از نظر جغرافیایی رخ می دهد، به اصطلاح ایزوله های ژنتیکی، این نسبت ها
نقض می شوند. نه تنها موانع جغرافیایی، بلکه موانع ملی، اجتماعی، مذهبی و غیره نیز می تواند منجر به انزوای ژنتیکی شود. جهش هایی که در اعضای جوامع بسته ایزوله ایجاد می شود در ایزوله های ژنتیکی گسترده تر می شود. این پدیده نامیده می شود اثر بنیانگذار. تغییر در بسامدهای آللی در طول یک سری از نسل ها می تواند به دلیل انتخاب تصادفی افراد که باعث ایجاد یک جمعیت یا بخشی از آن شده است، رخ دهد. این پدیده رانش ژنتیکی نامیده می شود. اثر بنیانگذار نوعی رانش ژنتیکی است. مهاجرت افراد می تواند با رانش ژنتیکی نیز همراه باشد.
همخونی باعث گسترش جهشهای خاص مرتبط با بیماریهای ارثی نادر در جدایههای ژنتیکی میشود. فراوانی برخی جهش ها در چنین جمعیت هایی می تواند چندین و گاه چندین ده برابر نسبت به سطح عمومی افزایش یابد. یک مثال کلاسیک که این نکات را نشان می دهد، گروه قومی یهودیان اروپای شرقی، به اصطلاح یهودیان اشکنازی است. در این گروه، فراوانی بیماریهای لیزوزومی نادر مانند بیماری گوچر،
تای ساکس (حماقت آموروتیک)، نیمن پیک، موکولیپیدوز، با
فرکانس های بالای دیستونی پیچشی، سندرم بلوم
(یکی از اشکال ژنتیکی کوتولگی، همراه با افزایش حساسیت به تابش خورشیدی، تلانژکتازی، اختلال در رنگدانه پوست و استعداد ابتلا به نئوپلاسم های بدخیم). علاوه بر این، افزایش فراوانی این بیماری ها، به عنوان یک قاعده، به دلیل توزیع گسترده جهش های خاص در ژن های مربوطه رخ می دهد. مثال دیگر شکل اتوزومال غالب بیماری پارکینسون است که در اثر جهش در ژن کیناز 2 غنی از لوسین - LRRK2 ایجاد می شود. در بیماران اروپایی مبتلا به اشکال خانوادگی بیماری، جهش اختصاصی در ژن LRRK2 (G2019S) با فراوانی 6 درصد رخ می دهد، در حالی که در همین بیماران، اما یهودیان اشکنازی، فراوانی این جهش به 30-40 درصد می رسد.
برخی جهشهای چند شکلی در دو ژن مرتبط با سرطان سینه و تخمدان در این گروه قومی شایع است. حتی یک بیماری شناخته شده مانند فیبروز کیستیک
یهودیان اشکنازی عمدتاً با وجود یک جهش بسیار خاص (W128X) توضیح داده می شوند. توجه داشته باشید که در اسرائیل، یک معاینه جامع از زنان باردار شامل تجزیه و تحلیل حامل هتروزیگوت جهش در برخی از ژن های مسئول بیماری های ذکر شده در بالا است.
طیف کاملاً متفاوتی از بیماری های ارثی که با افزایش فراوانی رخ می دهد در میان فنلاندی ها مشاهده می شود ، یعنی در یک گروه قومی دیگر. جهش های خاص فنلاندی برای حداقل 30 بیماری تک ژنی مختلف یافت شده است.
فراوانی نفروز مادرزادی در فنلاندی ها به 1:8000 می رسد. نزدیک
1% از جمعیت بومی فنلاند ناقل هتروزیگوت جهش هستند که در حالت هموزیگوت در بیش از
90٪ از بیماران مبتلا به دیسپلازی دیاستروفیک - یکی از اشکال دیسپلازی اسکلتی، که با اسکولیوز شدید، تغییر شکل مادرزادی دو طرفه دست و پا، ضخیم شدن گوش ها مشخص می شود.
کلسیفیکاسیون زودرس غضروف های دنده، وجود، در
در بیشتر موارد، شکاف کام سخت است. دو شکل از افتالموپاتی ارثی با افزایش فراوانی در میان فنلاندی ها رخ می دهد.
که هر کدام به دلیل جهش خاصی ایجاد می شوند. این آتروفی چین خورده مشیمیه و شبکیه و همچنین قرنیه مسطح نوع II است که در آن تیرگی قرنیه و پارانشیم قرنیه مشاهده می شود؛ پلاک های پیری دیسک بینایی یا صفحه زجاجیه در اوایل کودکی تشکیل می شوند.
در این حالت، سطح هایپرمتروپی به +10D یا حتی از آن فراتر می رود. با
لیپوفوسینوزیس سروئید نوزادی، آمیلوئیدوز خانوادگی، یکی از اشکال ژنتیکی، در جمعیت فنلاند با فراوانی افزایش یافته است.
صرع میوکلونیک پیشرونده (Unverricht-Lundborg).
نمونه هایی از این گونه ایزوله های ژنتیکی جدا نشده اند.
به طور کلی، شیوع جهش های مختلف در جمعیت ها بستگی به دو نیرو دارد که در جهت های مختلف عمل می کنند - فراوانی وقوع جهش ها و انتخاب منفی یا مثبت در رابطه با حامل های آنها. به عنوان مثال، جهشی که در حالت هموزیگوت تأثیر منفی بر زنده ماندن دارد، در صورتی که مزایایی در حالت هتروزیگوت داشته باشد، می تواند در جمعیت گسترده شود. مثال کلاسیک جهش در ژن β-گلوبین است که در صورت هموزیگوت منجر به کم خونی سلول داسی می شود. هموگلوبین جهش یافته حلالیت را کاهش داده و توانایی پلیمریزاسیون را افزایش داده است
در نتیجه گلبول های قرمز خون بیماران شکل داسی به خود می گیرند. چنین گلبول های قرمز خون انعطاف پذیری خود را از دست می دهند، عروق کوچک را مسدود می کنند و همولیز می شوند. سپس کانون های ایسکمی و انفارکتوس در اندام های داخلی، نخاع و مغز ایجاد می شود. این بیماری اغلب در آفریقای مرکزی، هند، کشورهای مدیترانه،
خاور نزدیک و میانه از جمله آذربایجان، ازبکستان و ارمنستان. معلوم شد که در همان مناطق جهان، پلاسمودیوم مالاریا، که باعث یک بیماری عفونی جدی - مالاریا می شود، گسترده است.
ناقلان هتروزیگتیک جهش در ژن β-گلوبین مقاومت به مالاریا را افزایش داده اند. فراوانی جهش های هتروزیگوت در ژن β-گلوبین در این جمعیت ها به 8-5 درصد می رسد.
مجموعه عوامل فوق منجر به چندشکلی جمعیت، یعنی همزیستی پایدار چندین فرم ژنتیکی در یک جمعیت، در حالی که جمعیت های مختلف ممکن است در سطوح یا فراوانی چندشکلی متفاوت باشند. ویژگی مهم یک فرد با یک ژنوتیپ خاص آن است
تناسب اندام (W)یعنی احتمال زنده ماندن تا سنین باروری و ترک فرزند. تناسب اندام جمعیت عمومی
مقدار متوسط تناسب اندام همه افراد و انحراف نرمال شده آن از حداکثر مقدار ممکن است - (Wmax -
W)/Wmax - تعیین می کند بار ژنتیکیجمعیت، که معیاری برای سنجش میزان شیوع در یک جمعیت جهش هایی است که تناسب اندام افراد را کاهش می دهد. این نسبت در کل جمعیت حامل های هموزیگوت و هتروزیگوت جهش هایی را تعیین می کند که تأثیر منفی بر زنده ماندن دارند. جمعیت های طبیعی گیاهان و جانوران و همچنین انسان ها با جهش های مختلفی مواجه هستند.
در رابطه با انسان، بار ژنتیکی میزان شیوع جهش های مرتبط با آسیب شناسی ارثی را در جمعیت های مختلف تعیین می کند. جهش های غالب به طور مداوم ظاهر می شوند
برخی جهشهای مغلوب در هموزیگوتهای نادر شناسایی میشوند، اما سهم اصلی بار ژنتیکی، مانند کوه یخ، در مخزن ژنی جمعیت در حالت هتروزیگوت پنهان است. به گفته ژنتیک برجسته روسی S.S.
جهش های Chetverikov در جمعیت های طبیعی ذخیره تکاملی گونه را تشکیل می دهند. این جهت از ژنتیک جمعیت در کشور ما در نیمه اول و اواسط قرن گذشته در آثار G. Meller، N.P. Dubinin و سپس R.L. Berg، M.D. Golubovsky و دیگران به شدت توسعه یافت. مشخص شد که غلظت جهش های مختلف در جمعیت ها، از جمله آنهایی که منجر به یک اثر کشنده در حالت هموزیگوت می شوند،
به چند ده درصد می رسد و ترکیب این جهش ها دائماً در حال تغییر است و جهش های مختلف خاص در سال های مختلف گسترده می شوند.
که در در خاتمه تاکید می کنیم کهژنتیکی جمعیت
انجام پژوهش از اهمیت بالایی برخوردار است
مطالعات اپیدمیولوژیک به منظور سازماندهی مناسب پزشکی
مشاوره ژنتیکی جمعیت و پیشگیری از آسیب شناسی ارثی
فصل 1.7. ساختار ماده وراثت - DNA
اختلالات عصبی و روانی 13 درصد از بار جهانی بیماری را تشکیل می دهند که به طور مستقیم بیش از 450 میلیون نفر در سراسر جهان را تحت تأثیر قرار می دهند. شیوع این اختلالات در نتیجه افزایش امید به زندگی در جمعیت احتمالاً همچنان در حال افزایش است. متأسفانه، تقریباً نیمی از بیماران مبتلا به اسکیزوفرنی در حال حاضر مراقبت های پزشکی مناسبی دریافت نمی کنند، تا حدی به این دلیل که علائم اولیه اسکیزوفرنی اغلب با علائمی که در سایر اختلالات روانی مشاهده می شود (مانند افسردگی روان پریشی یا اختلال دوقطبی) اشتباه گرفته می شود. سایر اختلالات مانند سندرم رت (RTT) و نوروفیبروماتوز نوع II (NF2) نیاز به رویکرد و درمان چند رشته ای در مراکز پزشکی تخصصی دارند. علاوه بر این، بیشتر این اختلالات پیچیده هستند که ناشی از تعامل عوامل ژنتیکی و محیطی است.
بر اساس داده های حاصل از مطالعات دوگانه، وراثت پذیری برخی از اختلالات روانی بالا است. این امر در مورد اوتیسم و اسکیزوفرنی با نرخ وراثت به ترتیب حدود 90٪ و 80٪ است. با این حال، این بیماریها اغلب بهعنوان موارد منفرد نیز رخ میدهند، و تنها یک کودک مبتلا از والدینی غیرمبتلا به دنیا میآید که سابقه خانوادگی ابتلا به این بیماری را ندارد. یکی از توضیحات احتمالی برای این پدیده، ظهور جهش است از نو، که در آن جهش در طی اسپرم زایی یا اووژنز (جهش های ژرملاین) رخ می دهد و بنابراین در بیمار وجود دارد اما در والد غیرمبتلا یافت نمی شود. این مکانیسم ژنتیکی اخیراً در تبیین بخشی از اساس ژنتیکی اختلالات رشدی سیستم مغزی مورد توجه قرار گرفته است.
با توجه به اینکه ژنوم انسان حدوداً 22333 ژن تخمین زده می شود، می توان گفت که بیش از 17800 ژن در مغز انسان بیان می شود. جهشهایی که تقریباً بر هر یک از این ژنها تأثیر میگذارند، در ترکیب با عوامل محیطی، میتوانند به ایجاد اختلالات عصبی و روانپزشکی مغز کمک کنند. مطالعات اخیر تعدادی از جهش های ژنی را شناسایی کرده و نقش مهمی را که ژنتیک در اختلالات عصبی و روانپزشکی ایفا می کند، نشان داده است. این مطالعات مشارکت نادر (<1% частоты) точечных мутаций и вариаций числа копий (CNVs, то есть геномных делеций или дублирования от>1 کیلوبایت تا چندین مگابایت) که ممکن است در مناطق عاری از ژن رخ دهد، یا ممکن است روی یک ژن تأثیر بگذارد، یا شامل مجموعهای از ژنها در علت ژنتیکی اوتیسم، اسکیزوفرنی، ناتوانی ذهنی، اختلال کمبود توجه و سایر اختلالات عصبی روانی باشد. .
مدتهاست که مشخص شده است که اختلالات عصبی و روانپزشکی در خانوادهها رخ میدهد که نشان دهنده وراثتپذیری با یک جزء ژنتیکی زمینهای برای این بیماری است. برای برخی از اختلالات عصبی، مانند NF2 یا RTT، یک علت ژنتیکی شناسایی شده است. با این حال، برای اکثریت قریب به اتفاق اختلالات عصبی و روانپزشکی، مانند اسکیزوفرنی، اوتیسم، اختلال دوقطبی و سندرم پای بیقرار، علل ژنتیکی تا حد زیادی ناشناخته باقی مانده است. پیشرفتهای اخیر در فناوریهای توالییابی DNA، فرصتهای جدیدی را برای درک ما از مکانیسمهای ژنتیکی زیربنای این اختلالات باز کرده است. با استفاده از پلت فرم های موازی توالی یابی DNA (که «نسل بعدی» نیز نامیده می شود)، می توان جهش ها را در تمام ژن های ژنوم یک فرد در یک نمونه (آزمایش) جستجو کرد.
ارزش شناخته شده د نووجهش (یعنی جهش اکتسابی در فرزندان) در اختلالات روانی مانند عقب ماندگی ذهنی (ID)، اوتیسم و اسکیزوفرنی. در واقع، در بسیاری از مطالعات ژنومی اخیر، تجزیه و تحلیل ژنوم افراد مبتلا و مقایسه آنها با ژنوم والدین نشان داده است که تغییرات کدگذاری و غیر کدگذاری نادر است. از نوبه طور قابل توجهی با خطر ابتلا به اوتیسم و اسکیزوفرنی مرتبط است. پیشنهاد شده است که تعداد زیادی از موارد جدید این اختلالات تا حدودی ناشی از جهش هستند نو،که ممکن است تلفات آللی را به دلیل کاهش شدید ظرفیت تولیدمثلی جبران کند و از این طریق فرکانس بالای این بیماری ها را حفظ کند. شگفت انگیز است که جهش ها از نوبسیار رایج هستند (به ترتیب 100 جهش جدید به ازای هر کودک)، با تنها تعداد کمی (به ترتیب یک کودک) در مناطق کدگذاری.
جهش ها از نوخارج از مناطق کد کننده، مانند نواحی پروموتر، اینترونیک، یا بین ژنی نیز ممکن است با بیماری همراه باشد. با این حال، چالش این است که مشخص شود کدام یک از این جهش ها بیماریزا هستند.
هنگام ارزیابی بیماری زایی یک مورد مشاهده شده باید چندین خط اصلی شواهد در نظر گرفته شود د نووجهش: د نوونرخ جهش، عملکرد ژن، تاثیر جهش، و همبستگی های بالینی. اکنون سؤالات کلیدی را می توان به صورت زیر فرموله کرد: چه تعداد ژن در اختلالات عصبی و روانی نقش دارند؟ چه مسیرهای ژنی خاصی درگیر هستند؟ عواقب جهش چیست؟ از نوبرای مشاوره ژنتیک؟ برای بهبود تشخیص و توسعه درمان باید به این سوالات پاسخ داده شود.
نقش جهش ها از نودر بیماری های انسانی، به ویژه در زمینه ژنتیک سرطان و اختلالات غالب مندلی مانند سندرم های کابوکی و شینزل-گیدیون به خوبی شناخته شده است. هر دوی این سندرم ها با ناتوانی ذهنی شدید و ناهنجاری های مادرزادی صورت مشخص می شوند و اخیراً نشان داده شده است که در اثر جهش ایجاد می شوند. از نو V ژن های MLL2و SETBP1، به ترتیب. اخیراً تحقیقات سندرز و همکاران.، نیل و همکاران.، اورواک و همکاران. مشارکت را تایید کرد د نووجهش در علت اوتیسم هر مطالعه فهرستی از جهش ها را شناسایی کرد نو،در پروباندها وجود دارد، اما تنها چند ژن با چندین مورد شناسایی شده است از نو (CHD8، SCN2A، KATNAL2و NTNG1). برهمکنش پروتئین و آنالیزهای مبتنی بر مسیر از این مطالعات، ارتباط قابل توجهی و یک مسیر بیولوژیکی مشترک بین ژنهای حامل جهش را نشان داد. از نودر موارد اوتیسم شبکه های پروتئینی درگیر در بازسازی کروماتین، یوبی کوئیتیناسیون و رشد عصبی به عنوان اهداف بالقوه ژن های حساسیت به اوتیسم شناسایی شده اند. در نهایت، این مطالعات نشان می دهد که 1000 یا بیشتر ژن را می توان به عنوان احتمال داشتن جهش های فراگیر که در ایجاد اوتیسم نقش دارند، تفسیر کرد.
پیشرفتهای تکنولوژیکی در توالییابی DNA اساساً مطالعه تغییرات ژنتیکی در ژنوم انسان را متحول کرده است و شناسایی بسیاری از انواع جهشها، از جمله جانشینیهای تک جفت باز، درجها/حذفها، CNVs، وارونگیها و بسطهای تکراری و همچنین مواردی که در نظر گرفته شدهاند را ممکن ساخته است. جهش های سوماتیک و ژرمینال نشان داده شده است که همه این نوع جهش ها در بیماری انسان نقش دارند. به نظر میرسد جهشهای تک نوکلئوتیدی عمدتاً «منشا پدری» دارند، در حالی که حذفها ممکن است در درجه اول «منشا مادری» داشته باشند. این ممکن است با تفاوت بین گامتوژنز نر و ماده توضیح داده شود. به عنوان مثال، در مطالعه ای در مورد نوروفیبروماتوز، 16 جهش از 21 جهش شامل حذف هایی با منشاء مادری بود و 9 مورد از 11 جهش نقطه ای منشأ پدری داشتند.
انواع مختلفی از جهش ها می توانند از والدین به فرزند منتقل شوند یا به طور خود به خود به دست آیند. مکانیسم محرک دومی در سال های اخیر به دلیل اهمیت این نوع جهش در بیماری هایی مانند اسکیزوفرنی و اوتیسم مورد توجه قرار گرفته است. میزان جهش نو،به نظر می رسد با سن پدر غالب است. این میزان در اینجا با افزایش سن پدر افزایش مییابد، احتمالاً به دلیل پیامدهای کاهش بازده تکثیر DNA یا مکانیسمهای ترمیم، که انتظار میرود با افزایش سن کاهش یابد. بنابراین خطر ابتلا به این بیماری باید با افزایش سن پدر افزایش یابد. مشخص شده است که این در بسیاری از موارد رخ می دهد، از جمله سندرم کروزون، نئوپلازی غدد درون ریز متعدد نوع II، و نوروفیبروماتوز نوع I. اخیراً، O'Roak و همکاران. یک مولفه قابل توجه پدری را در 51 جهش مشاهده کرد نو،از طریق مطالعه توالی 188 والدین-کودکان مبتلا به اوتیسم پراکنده شناسایی شد. این نتایج مشابه نتایجی است که در گزارش های اخیر مشاهده شده است CNN n نووبا ناتوانی ذهنی این همبستگی ممکن است با تعداد قابل توجهی بیشتر تقسیم سلولی میتوز در سلولهای زایا یا اسپرماتوسیتها قبل از میوز در طول عمر مردان در مقایسه با آنچه در طول اووژنز در زنان رخ میدهد توضیح داده شود.
بر اساس تعداد ثابت تقسیمات سلولی که در اووژنز (از بدو تولد تا یائسگی) در مقایسه با اسپرماتوژنز (از بلوغ تا پایان زندگی) رخ می دهد، جیمز اف. کرو تخمین زد که در سن 30 سالگی میانگین تعداد کروموزوم ها از زیگوت به اسپرم تکرار می شود. تولید 16.5 برابر بیشتر از زیگوت تا تشکیل تخم است.
موزاییک ژنتیکی ناشی از وقوع است از نوجهشهای میتوزی، خود را خیلی زود در رشد جنینی نشان میدهند و به عنوان وجود کلونهای سلولی متعدد با یک ژنوتیپ خاص در یک فرد تعریف میشود. موزائیسم سوماتیک و ژرمینی وجود دارد، اما موزاییک ژرمینی ممکن است در انتقال آنچه می تواند توسط جهش منتقل شود کمک کند. از نوآیندگان
جهشهای خودبهخودی که در سلولهای بدنی (در حین میتوز، پس از لقاح) رخ میدهند نیز میتوانند در پیدایش بیماریهای مرتبط با اختلالات رشدی نقش داشته باشند.